") 23.12.2022
23.12.2022
Рак коры надпочечника (Адренокортикальный рак)
Первичный гиперальдостеронизм - клинический синдром, развивающийся в результате избыточной продукции альдостерона клубочковой зоной коркового вещества надпочечников, при котором секреция альдостерона полностью или частично автономна по отношению к ренин-ангиотензиновой системе
• Общероссийский национальный союз "Ассоциация онкологов России"
• • Общественная организация "Российская Ассоциация Эндокринологов"
Одобрено Научно-практическим Советом Минздрава РФ
Список сокращений
18ФДГ-ПЭТ/КТ - позитронно-эмиссионная томография с 18-фтордезоксиглюкозой, совмещенная с компьютерной томографией
AJCC - Американский объединенный комитет по раку
ENSAT - Европейская рабочая группа по изучению опухолей надпочечников (European Network for the Study of Adrenal Tumors)
HU (единицы Хаунсфилда) - денситометрические показатели при проведении КТ
RECIST - Критерии ответа солидных опухолей на терапию (Response evaluation criteria in solid tumors)
SUV (standartised uptake value) - накопительный критерий при 18ФДЕ-ПЭТ/КТ
TNM - Международная классификация стадий злокачественных новообразований (аббревиатура от Tumor, Nodus, Metastasis)
UICC - Международный союз по борьбе с раком (Union for International Cancer Control)
АГ - артериальная гипертензия
АКР - рак коры надпочечника (син.: адренокортикальный рак)
АКТГ - адренокортикотропный гормон
АЛТ - аланин-амминотрансфераза
АРС - альдостерон-рениновое соотношение
ACT - аспартат-аминотрансфераза
ВОЗ - Всемирная организация здравоохранения
дат - дистанционная лучевая терапия
ггтп - гамма-глутамил-транспептидаза
ГСПГ - глобулин, связывающий половые стероиды
ИГХ - иммуногистохимическое исследование
КУ - контрастное усиление
МКБ-10 - Международная классификация болезней 10-го пересмотра
МРТ - магнитно-резонансная томография
МСКТ - мультиспиральная компьютерная томография
МЭН - синдром множественных эндокринных неоплазий
нпв - нижняя полая вена
ОБП - органы брюшной полости
ОГК - органы грудной клетки
ПГА - первичный гиперальдостеронизм
ФХЦ/ПГ - феохромоцитома/параганглиома
СИК - синдром Иценко-Кушинга
ТТГ - тиреотропный гормон
Термины и определения
Адъювантная химиотерапия - химиотерапия, применяемая после локального воздействия на опухоль в целях эрадикации или длительного подавления микрометастазов
Герминативная мутация - изменение структуры гена (последовательности нуклеотидов, хромосомы, генома), по сравнению с референсной последовательностью, возникшее в половых (зародышевых) клетках.
Гиперкортицизм (эндогенный) - комплекс клинических симптомов, обусловленных длительным воздействием кортикостероидов на организм вследствие их избыточной эндогенной продукции.
Драйверная мутация - изменение структуры гена (последовательности нуклеотидов, хромосомы, генома), инициирующее превращение нормальной клетки в раковую
Инциденталома надпочечника - собирательное понятие, включающее разнообразную по морфологии группу опухолей более 1 см в диаметре, случайно выявленных при радиологическом обследовании.
Канцерогенез - сложный патофизиологический процесс зарождения и развития опухоли.
Мутация - стойкое (то есть такое, которое может быть унаследовано потомками данной клетки или организма в случае герминативной мутации) изменение генома.
Острая надпочечниковая недостаточность - симптомокомплекс, обусловленный резким снижением или полным прекращением функциональной деятельности коры надпочечников.
Орфанное заболевание - редкое заболевание, которое встречается у небольшого количества людей относительно общей численности населения: в Европе редким принято считать заболевание с распространенностью 1 человек на 2000 населения, в СТТТА — если затрагивают не менее 2000 человек, в России — не более 10 человек на 100 000 населения.
Первичный гиперальдостеронизм - клинический синдром, развивающийся в результате избыточной продукции альдостерона клубочковой зоной коркового вещества надпочечников, при котором секреция альдостерона полностью или частично автономна по отношению к ренин-ангиотензиновой системе, что обусловливает развитие низкорениновой гипокалиемической артериальной гипертензии.
Сигнальный путь - последовательность молекул, посредством которых информация от клеточного рецептора передается внутри клетки.
Синдром «неуправляемой гемодинамики» - патологическое состояние, возникающее на фоне катехоламинпродуцирующей опухоли надпочечника и характеризующееся резким учащением гипертензивных высокоамплитудных кризов с усугубляющейся некурабельной гипотонией в межприступном периоде
Системная химиотерапия - введение препаратов внутрь, подкожно, внутривенно, внутримышечно, рассчитанное на общий противоопухолевый эффект.
Феохромоцитома - опухоль хромаффинных клеток симпато-адреналовой системы надпочечниковой или вненадпочечниковой локализации, как правило, секретирующая катехоламины.
Термины и определения
Адъювантная химиотерапия - химиотерапия, применяемая после локального воздействия на опухоль в целях эрадикации или длительного подавления микрометастазов
Герминативная мутация - изменение структуры гена (последовательности нуклеотидов, хромосомы, генома), по сравнению с референсной последовательностью, возникшее в половых (зародышевых) клетках.
Гиперкортицизм (эндогенный) - комплекс клинических симптомов, обусловленных длительным воздействием кортикостероидов на организм вследствие их избыточной эндогенной продукции.
Драйверная мутация - изменение структуры гена (последовательности нуклеотидов, хромосомы, генома), инициирующее превращение нормальной клетки в раковую
Инциденталома надпочечника - собирательное понятие, включающее разнообразную по морфологии группу опухолей более 1 см в диаметре, случайно выявленных при радиологическом обследовании.
Канцерогенез - сложный патофизиологический процесс зарождения и развития опухоли.
Мутация - стойкое (то есть такое, которое может быть унаследовано потомками данной клетки или организма в случае герминативной мутации) изменение генома.
Острая надпочечниковая недостаточность - симптомокомплекс, обусловленный резким снижением или полным прекращением функциональной деятельности коры надпочечников.
Орфанное заболевание - редкое заболевание, которое встречается у небольшого количества людей относительно общей численности населения: в Европе редким принято считать заболевание с распространенностью 1 человек на 2000 населения, в СТТТА — если затрагивают не менее 2000 человек, в России — не более 10 человек на 100 000 населения.
Первичный гиперальдостеронизм - клинический синдром, развивающийся в результате избыточной продукции альдостерона клубочковой зоной коркового вещества надпочечников, при котором секреция альдостерона полностью или частично автономна по отношению к ренин-ангиотензиновой системе, что обусловливает развитие
низкорениновой гипокалиемической артериальной гипертензии.
Сигнальный путь - последовательность молекул, посредством которых информация от клеточного рецептора передается внутри клетки.
Синдром «неуправляемой гемодинамики» - патологическое состояние, возникающее на фоне катехоламинпродуцирующей опухоли надпочечника и характеризующееся резким учащением гипертензивных высокоамплитудных кризов с усугубляющейся некурабельной гипотонией в межприступном периоде
Системная химиотерапия - введение препаратов внутрь, подкожно, внутривенно, внутримышечно, рассчитанное на общий противоопухолевый эффект.
Феохромоцитома - опухоль хромаффинных клеток симпато-адреналовой системы надпочечниковой или вненадпочечниковой локализации, как правило, секретирующая катехоламины.
1. Краткая информация
1.1 Определение заболевания или состояния (группы заболеваний или состояний)
Рак коры надпочечника (синадренокортикальный рак; АКР) - редкая злокачественная опухоль коры надпочечников, характеризующая, как правило, поздним сроком выявления, агрессивностью клинического течения и неблагоприятным лечебным прогнозом [1].
1.2 Этиология и патогенез заболевания или состояния (группы заболеваний или состояний)
Канцерогенез при АКР обусловлен драйверными мутациями ряда генов и активацией соответствующих сигнальных путей [2]. В большинстве случаев эти мутации возникают спонтанно в соматических клетках коры надпочечника, приводя к спорадическим случаям заболевания. Также в основе патогенеза АКР могут лежать герминативные мутации: в этом случае заболевание будет наблюдаться в рамках того или иного наследственного синдрома [3].
Описано несколько наследственных синдромов, компонентом которых является АКР (Табл. 1). В рамках всех синдромов отмечается низкая фенотипическая пенетрантность АКР, в связи с чем, проследить наследственный характер заболевания анамнестически достаточно сложно [1]. Самым распространенным наследственным синдромом, включающим АКР, является синдром Ли-Фраумени, обнаруживаемый в большинстве случаев АКР в детском возрасте - до 80% всех пациентов; он также является самой частой причиной наследственного АКР во взрослом возрасте - до 5% всех случаев заболевания. Распространенность остальных синдромов, ассоциированных с АКР, составляет от долей процента до 3-4% среди взрослых пациентов с АКР [3].
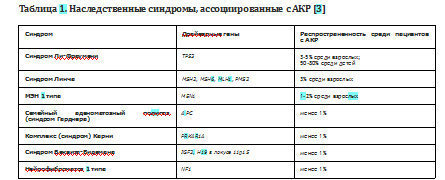
В основе патогенеза синдрома Линча лежат герминативные мутации в одном из генов системы репарации ДНК (MSH2, MSH6, MLH1, PMS2), что приводит к развитию рака проксимальных отделов толстой кишки (до 80% случаев синдрома Линча). Помимо колоректального рака и АКР, у пациентов с синдромом Линча повышены риски злокачественных образований других локализаций: рака эндометрия (50-71% случаев синдрома Линча), почечной лоханки, мочевого пузыря, мочеточников, яичников, желудка, тонкой кишки, поджелудочной железы, а также глиобластом головного мозга и опухолей сальных желез. Могут наблюдаться такие кожные проявления, как кератоакантомы и пятна цвета «кофе с молоком».
Синдром множественных эндокринных неоплазий (МЭН) 1 типа обусловлен герминативными мутациями в гене-супрессоре опухолевого роста MEN1. Наиболее распространенным фенотипом адренокортикальных поражений, наблюдаемым при синдроме МЭН 1 типа, являются односторонняя или двусторонняя гиперплазия коры надпочечников и аденомы.
Эти поражения встречаются у 45-55% пациентов с МЭН 1 и могут быть гормонально-активными или нефункциональными. Распространенность АКР в рамках синдрома МЭН 1 типа достигает 22%. Другими проявлениями синдрома МЭН 1 типа наиболее часто являются гиперплазия или опухоли околощитовидных желез, приводящие к первичному гиперпаратиреозу, панкреатические нейроэндокринные опухоли, опухоли гипофиза.
АКР также может выявляться в составе синдромов Гарднера (аденоматозный полипоз кишки), Беквита-Видемана, комплекса Карни, нейрофиброматоз 1 типа. Доля этих синдромов в структуре АКР, по данным литературы, не превышает 1% [3,4].
1.3 Эпидемиология заболевания или состояния (группы заболеваний или состояний)
Ежегодное выявление АКР составляет 0,5-2 случая на 1 миллион населения, в структуре онкологической смертности составляет 0,04-0,2%. Наибольшая заболеваемость наблюдается в южных регионах Бразилии, что объясняется высокой распространенностью герминативной мутации R337H гена ТР53.
На момент постановки диагноза средний возраст пациента составляет 50-60 лет, однако заболевание также встречается у детей. Женщины болеют чаще, соотношение женщины/ мужчины составляет 2,5:1 [2; 5].
Эпидемиологические данные по Российской Федерации оценить крайне затруднительно, так как онкологическая отчетность отдельно по АКР не формируется, регистр отсутствует.
АКР включен в перечень орфанных заболеваний в РФ [6].
1.4 Особенности кодирования заболевания или состония (группы заболеваний или состояний) по Международной статистической классификации болезней и проблем, связанных со здоровьем
С74 Злокачественное новообразование надпочечника С74.0 Коры налпочечника С74.9 Надпочечника неуточненной части
1.5 Классификация заболевания или состояния (группы заболеваний или состояний)
1.5.1. Международная гистологическая классификация
Согласно Международной гистологической (классификация Всемирной организации здравоохранения (ВОЗ), 4-е издание, 2017 г.), АКР кодируется как 8370/3 (Табл. 2) [2].


1.5.2. Стадирование
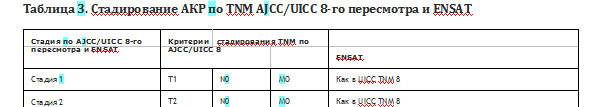
Стадирование АКР проводят по классификации TNM Американского объединенного комитета по раку (А1СС)/Союза международного контроля над раком (UICC) 8-го пересмотра (2017 г.) и по классификации Европейской рабочей группы по изучению опухолей надпочечников (ENSAT, European Network for the Study of Adrenal Tumors) [7;8].
Клиническое стадирование АКР осуществляется на предоперационном этапе на основании результатов визуализирующих диагностических методов. После проведения хирургического лечения для надлежащего стадирования АКР необходимо гистологическое подтверждение (pTNM).
Первичная опухоль (Т/рТ). Критерий Т отражает распространенность первичной опухоли и содержит следующие градации:
Тх: Первичная опухоль не может быть оценена
ТО: нет данных о наличии первичной опухоли
Т1: размер опухоли < 5 см;
Т2: размер опухоли > 5 см;
ТЗ: опухоль любого размера с локальной инвазией, без вовлечения окружающих органов*;
Т4: опухоль любого размера с инвазий опухоли в окружающие органы и/или тромбоз нижней полой вены (НПВ) и/или тромбоз почечной вены.
* Окружающими органами являются: почка, диафрагма, крупные сосуды, поджелудочная железа, печень.
Регионарные лимфатические узлы (N/pN). Критерий N указывает на наличие или отсутствие метастазов в регионарных лимфатических узлах
Nx: регионарные лимфоузлы не могут быть оценены;
N0: отсутствие метастатического поражения лимфоузлов;
N1: метастатическое поражение лимфоузлов
Регионарными считаются лимфоузлы ворот почки, парааортальные и паракавальные. Сторона поражения надпочечника не учитывается.
Отдаленные метастазы (М). Критерий М характеризует наличие или отсутствие отдаленных метастазов:
МО: отдаленных метастазов отсутствуют
Ml: отдаленные метастазы присутствуют
Группировка критериев TNM для определения стадии АКР по UICC и ENSAT представлена в таблице 3. Критерии установления стадии АКР идентичны как для UICC, так и для ENSAT.

1.6 Клиническая картина заболевания или состояния (группы заболеваний или состояний)
АКР может быть гормонально-активным, являясь, чаще всего, причиной субклинического или манифестного синдрома Иценко-Кушинга (СИК), и/или вирильного синдрома, или может быть случайно выявленной гормонально-неактивной опухолью надпочечника
Клинические проявления АКР в разных возрастных группах несколько отличаются.
У взрослых пациентов клинические проявления АКР включают симптомы избыточной гормональной секреции (50-60%) и неспецифические симптомы, связанные с ростом опухоли (30-35%) [2;9;10]. В остальных случаях (10-15%) заболевание обнаруживается случайно при проведении визуализирующих исследований: мультиспиральной компьютерной томографии (МСКТ) и/или магнитно-резонансной томографии (МРТ) и/или ультразвукового исследования (УЗИ) как инциденталома (Табл.4) [8-13].

Изолированная гиперпродукция андрогенов является вторым по распространенности проявлением гормональной активности АКР (до 20% пациентов). Гиперандрогенемия у женщин может приводить к развитию гирсутизма, вирилизации и нарушению менструального цикла. У мужчин избыток андрогенов часто остается незамеченным ввиду скудности симптомов. Гиперпродукция эстрогенов при АКР встречается редко (менее 2%), вызывает у мужчин развитие гинекомастии и атрофии яичек, у женщин в постменопаузе проявляется метроррагиями. Избыточная секреция половых гормонов при обнаружении опухоли надпочечника особенно подозрительна в отношении АКР.
Наиболее редкий вариант эндокринопатии при АКР связан с проявлениями первичного гиперальдостеронизма, который манифестирует выраженной артериальной гипертензией и гипокалиемией. Повышенная продукция альдостерона отмечается несколько чаще в рамках смешанной гормональной продукции.
В большинстве случаев АКР имеет относительно большие размеры, в среднем - более 10 см; опухоль может увеличиваться до 25 см и более. Неспецифические симптомы АКР, такие как дискомфорт или боли, боль в поясничной и абдоминальной областях, ощущение переполнения желудка, обусловлены ростом опухоли в размерах и компрессией прилежащих органов [14].
Симптомы, традиционно ассоциируемые с онкологическим заболеванием (похудение, повышенное потоотделение в ночное время, выраженная слабость, лихорадка), как правило, не характерны для АКР [8].
У детей гормональная активность АКР выявляется чаще, чем у взрослых, и составляет 87-95% случаев заболевания. Большинство опухолей изолированно секретирует кортизол (65%) или андрогены в комбинации с кортизолом (30%). Другие варианты гормональной активности в детском возрасте можно считать казуистическими.
2. Диагностика
1. жалобы пациента;
2. данные индивидуального и семейного анамнеза;
3. результаты физикального обследования;
4. результаты лабораторных, инструментальных и иных диагностических исследований.
Диагностика неметастатического АКР осуществляется в случае выявления опухоли надпочечника. С этой точки зрения диагностика АКР рассматривается в рамках дифференциального диагноза с другими опухолями надпочечников, наиболее часто выявляемыми случайно (инциденталомами). Термин «инциденталома надпочечника» является собирательным, включающим разнообразную по морфологии группу опухолей более 1 см в диаметре, случайно выявленных при радиологическом обследовании [15].
Выявленное образование может
• оказаться как гормонально-неактивным, так и активно производить различные гормоны;
• исходить из различных зон надпочечника или иметь неспецифичную органную принадлежность;
• быть злокачественным или доброкачественным.
Диагностика местнораспрострненного или метастатического рака как правило не вызывает трудностей, иногда проводится дифференциальная диагностика с опухолями печени, почки, забрюшинными неорганными опухолями.
При АКР небольших размеров с массивным отдаленным метастатическим поражением может возникнуть необходимость дифференциальной диагностики с другой злокачественной опухолью с метастазом в надпочечник
(например, рака легкого).
2.1 Жалобы и анамнез
Жалобы пациентов с гормонально-активным АКР определяются характером секреторного спектра опухоли:
• гиперкортицизм: общая слабость, головные боли, диспластичное ожирение, артериальная гипертензия, мышечная слабость, матронизм, «климактерический горбик», яркие широкие стрии (передняя брюшная стенка, бедра, подмышечные области и т.д.), нарушения менструального цикла, снижение полового влечения, сахарный диабет, и др.;
• гиперандрогенемия: у женщин гирсутизм, нарушения менструального цикла, увеличение клитора; повышение жирности и нечистота кожи;
• гиперэстрогенемия: импотенция, гинекомастия у мужчин, метроррагии у женщин в постменопаузе;
• гиперальдостеронизм: артериальная гипертензия, не корригируемая многокомпонентной терапией, мышечная слабость.
Гормонально-неактивные АКР могут проявляться неспецифическими жалобами на дискомфорт/болями в животе или спине, ощущение переполнения в животе, наличие объемного образования в брюшной полости, определяемого самостоятельно при ощупывании живота.
В 10-15% всех случаев пациенты с АКР не предъявляют жалоб, в этом случае АКР первично выявляется как инциденталома.
2.2 Физикальное обследовани
Проявления АКР, выявляемые при физикальном обследовании определяются симптоматикой гормональной гиперпродукии (см. разделы 1.6 и 2.1). Опухоль больших размеров может быть обнаружена пальпаторно. Возможно диагностирование лихорадки вследствие сопутствующего некроза в опухоли. Клинические проявления могут иметь метастазы АКР в печень, легкие, кости.
2.3 Лабораторные диагностические исследования
• 2.3.1. Рекомендуется всем пациентам при выявлении опухоли надпочечника размером более 1 см в первую очередь лабораторные исследования для исключения или подтверждения гормональной активности образования, которая может проявляться гиперкатехоламинемией, АКТГ-независимым гиперкортицизмом, первичным гиперальдостеронизмом [13,17].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 5
Комментарий. По данным сводной аутопсийной статистики, распространенность случайно выявленных опухолей надпочечника составляет в среднем - 6%. По данным МСКТ, “•случайные” образования надпочечника выявляются приблизительно у 4% обследованных пациентов [16]. В возрасте до 30 лет инциденталома встречается приблизительно у 0,2% обследованных, но в группе пациентов старше 70 лет частота возрастает до 7%>.
Представления о частоте выявления АКР при инциденталомах пересмотрена в связи с расширением клинической базы исследований и требованиями к характеру и дизайну эпидемиологических исследований. Так, по данным W.F. Young и соавт. [17], частота АКР оценивалась чуть более 4%. Позднее, межклинический анализ [13] большого числа пациентов продемонстрировал распространенность АКР среди инциденталом не более 1,9%.
• 2.3.2. Исследование гормональной активности опухоли надпочечника строго регламентировано и рекомендуется учитывать его результаты для каждого пациента при планировании предоперационной подготовки, объема операции, последующего наблюдения [13,24].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 5
Комментарий. Отсутствие любых клинических проявлений и манифестных признаков гормональной активности является, к сожалению, недостаточно известной, но довольно распространенной “ловушкой ”.
В связи с увеличивающейся распространенностью и доступностью методов топической диагностики, начиная с двух последних декад прошлого века, отмечается увеличение числа инциденталом надпочечника. Это, в свою очередь, привело к увеличению удельного веса клинически «немых» феохромоцитом/параганглиом (ФХЦ/ПГ) с 10 до 40-45%. При случайно выявленных опухолях частота встречаемости ФХЦ/ПГ оценивается в 5-6%, при этом клинически “немые” формы составляют подавляющее большинство. Достаточно часто выявляется феномен субклинического гиперкортицизма - 8-12% наблюдений среди инциденталом [18,19].
Иногда встречается обратная ситуация: яркая клиническая картина гиперпродукции определенного гормона нацеливает врача на “очевидный” диагноз, казалось бы, не требующий тщательной гормональной диагностики.
Зачастую, морфологически опухоль имеет иную природу, отличную от “очевидного” диагноза. Так, в литературе неоднократно описаны ФХЦ/ ПТ, проявляющиеся не только гиперкатехоламинемией, но и эктопированной продукцией кортикотропина, проявляющегося «большими» признаками гиперкортицизма [20-22].
Описаны протекающие с классическими гипертензивными кризами «ФХЦ/ПГ», которые оказываются гормонально-неактивными аденомами коры надпочечника, или, что гораздо хуже -АКР [23]. •
• 2.3.3.1 Рекомендуется всем пациентам с выявленной опухолью надпочечника проводить исследование уровня общего кортизола в кровиранние утренние часы на фоне подавляющего теста с 1 мг дексаметазона с целью верификации гиперкортицизма [19, 24, 27]. Недооценка наличия гиперкортицизма (манифестного или субклинического) связана с высоким риском развития острой надпочечниковой недостаточности в послеоперационном периоде.
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 5
• 2.3.3.2. При отсутствии физиологического подавления уровня кортизола в качестве подтверждающего теста рекомендуется использовать исследование уровня адренокортикотропного гормона в крови (АКТГ) в утренние часы [13,17,24].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 5
Комментарий. Пациенты с инциденталомой надпочечника редко имеют развернутую клиническую картину гинеркортицизма, однако биохимические признаки гиперкортицизма отмечаются примерно у 8-12% пациентов [24].
У здоровых лиц назначение супрафизиологической дозы глюкокортикоидов проявляется подавлением АКТГ и синтеза кортизола. При назначении низких доз синтетического глюкокортикоида дексаметазона при синдроме эндогенного гиперкортицизма любого генеза этого подавления не происходит [25]. Ночной тест достаточно прост и выполним в амбулаторных условиях: 1 мг дексаметазона принимается между 23 и 24 часами, кортизол измеряется в крови, взятой следующим утром между 8 и 9 часами. Более высокие дозы (1,5 или 2 мг) не улучшают точность теста [26].
В качестве диагностического критерия с высокой отрицательной прогнозирующей ценностью в отношении субклинического гиперкортицизма предложено считать подавление утреннего кортизола менее 50 нмоль/л (чувствительность более 95%) [24-26].
При инциденталомах надпочечников определение суточного кортизола мочи обладает меныией чувствительностью в сравнении с дексаметазоновым тестом и ночным сывороточным кортизолом [27, 28]. Однако специфичность подавляющего теста при уровне cut-off менее 50 нмоль/л не превышает 80%. Для уменьшения числа ложноположительных результатов в качестве верифицирующего теста используется определение в утренние часы уровня АКТГ. Подавленный уровень АКТГ подтверждает диагноз синдрома Иценко-Кушинга у пациентов с опухолями надпочечников [28]. Измерение АКТГ не является методом первичной диагностики, однако может служить подтверждающим признаком субклинических проявлений гиперкортицизма в этой группе пациентов.
Возникновение послеоперационной острой надпочечниковой недостаточности является основной причиной, из-за которой диагностика субклинического гиперкортицизма является строго обязательной. Клинические проявления являются неспецифическими для послеоперационного периода и часто мимикрируют под другие осложнения (кровотечение, интоксикация, острый инфаркт миокарда и т.д.): отсутствие аппетита, слабость, тошнота, умеренная гипотония, вздутие живота, вечерние гектические подъемы температуры. Несвоевременно распознанные и не купированные, эти клинические проявления могут привести к фатальному исходу [29].
Опухоль коры надпочечника, автономно продуцирующая кортизол, является причиной атрофии коры контрлатерального надпочечника. Если при манифестном синдроме Иценко-Кушинга можно оценить продолжительность гиперкортицизма по давности клинических проявлений, то при субклинических формах его длительность неизвестна. При длительном течении гиперкортицизма возможна необратимая атрофия коры, требующая длительной, если не пожизненной, заместительной терапии [30].
• 2.3.4 Рекомендуется всем пациентам с выявленной опухолью надпочечника проводить исследование уровней метанефрина и норметанефрина в крови или в суточной порции мочи с целью верификации ФХЦ/ПГ [13, 31, 32, 33]. Недооценка наличия катехоламин-секретирующей опухоли связана с высоким риском периоперационного развития высокоамплитудных гипертензивных кризов, отека легких, фатальных аритмий, синдрома “неуправляемой гемодинамики” и внезапной сердечной смерти.
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 4
Комментарий. Даже если клиническое течение ФХЦ/ПГ было “бессимптомным”, высокий уровень циркулирующих катехоламинов во время операции приводит к гипертензивным кризам, аритмиям, синдрому “неуправляемой гемодинамики” [30]. Под последним понятием подразумевается резкое учащение гипертензивных высокоамплитудных кризов с усугубляющейся некурабельной гипотонией в межприступном периоде.
К серьезным лечебным ошибкам приводит недооценка гиповолемического синдрома у пациентов с ФХЦ/ПГ. При снижении объема циркулирующей жидкости и выраженной вазоконстрикции, определяемое на периферии АД будет значительно ниже, чем при нормоволемии. Отсутствие высокого артериального давления на периферии сопровождается гипертензией в центральных сосудах [31 ].
С другой стороны, длительное бессимптомное течение может быть объяснено внутриопухолевым метилированием активных фракций катехоламинов опухолевым ферментом катехоламин-О-метил-трансферазой (КОМТ).
Однако, интраоперационно, при механическом давлении на опухоль (при пневмоперитонеуме, манипуляциях) или при фармакологической провокации интраоперационный выброс активных фракций может обуславливать дебютную симптоматику ФХЦ/ПГ. В связи с этим всем пациентам с биохимически подтвержденной ФХЦ/ПГ необходимо проводить предоперационную подготовку, целью которой является нивелирование воздействия циркулирующих катехоламинов на адренорецепторы [32-34].
• 2.3.5 Рекомендуется всем пациентам с опухолью надпочечника при наличии артериальной гипертензии проводить определение соотношения между уровнем альдостерона и активностью ренина плазмы крови для исключения первичного гиперальдостеронизма [36,37].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 5
Комментарий. Изолированная автономная гиперпродукция альдостерона при АКР встречается крайне редко. Альдостерон-продуцирующие опухоли морфологически выглядят как светлоклеточные аденомы небольших размеров [16]. Тестом первичной диагностики является оценка альдостерон-ренинового соотношение (АРС). При подозрении на первичный гиперальдостеронизм (ПГА) по результатам АРС, необходимо проведение одного из подтверждающих методов. Алгоритм обследования при ПГА подробно изложен в клинических рекомендациях по диагностике и лечению данной нозологии [35, 36].
• 2.3.6 Рекомендуется всем пациентам с опухолью надпочечника при клиническом подозрении на изолированную или сочетанную с гиперкортицизмом опухолевую гиперпродукцию половых гормонов проводить комплексное определение концентрации стероидных гормонов в крови: дегидроэпиандростерона сульфата, 17- гидроксипрогестерона, андростендиона, общего тестостерона (у женщин), общего эстрадиола (у мужчин и женщин в менопаузе), 11 - дезоксикортикостерона [8;]
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 5
Комментарий. Стероидный профиль пациента с опухолью надпочечника может указывать на злокачественную природу образования. Сочетанная автономная гиперпродукция андрогенов и кортизола у пациентов обоего пола, секреция предшественников стероидов или эстрадиола у мужчин говорят в пользу высоковероятного диагноза АКР [129].
Напротив, умеренно выраженный, длительный гирсутизм на фоне гиперандрогенемии у женщин более вероятно обусловлен синдромом поликистозных яичников или неклассической формой врожденной дисфункции коры надпочечников [130].
2.4 Инструментальные диагностические исследования
• 2.4.1. Рекомендуется всем пациентам с опухолью надпочечника для диагностики злокачественного потенциала опухоли провести оценку количественных денситометрических показателей при трехфазной КТ органов брюшной полости с контрастным усилением (КУ) при отсутствии абсолютных противопоказаний к проведению КТ:
о плотность тканевого компонента до контрастирования (нативная); о плотность в тканевую фазу контрастирования (артериальная и венозная фазы); о плотность в отсроченную (через 10 мин. после введения контраста) фазу контрастирования (фаза вымывания).
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 5
Комментарий. При получении высокоплотных КТ-значений в нативную фазу, задержки контраста в отсроченной фазе - злокачественный потенциал опухоли должен оцениваться, как высокий. Основным критерием при формировании показаний к операции долгое время являлся размер новообразования, что приводило, с одной стороны, к большому числу необоснованных вмешательств при опухолях более 3-4 см, с другой стороны - к недооценке злокачественного потенциала образований малого размера при метастатических поражениях и АКР. При изучении итальянского регистра [37, 38] лишь 25% опухолей надпочечников более 4 см оказались злокачественными. Одновременно авторы привели данные, что среди пациентов с АКР опухоли менее 6 см составляли около 10% наблюдений; с другой стороны, среди всех опухолей надпочечника более 6 см в диаметре АКР составил только 20% (!). В более поздних исследованиях, при введении денситометрических критериев злокачественности опухоли, удалось оптимизировать показания к оперативному лечению не только на основании размеров опухоли. В настоящий момент основное внимание в диагностике АКР сконцентрировано на количественных показателях трехфазной КТ.
КТ-семиотика инциденталом надпочечника оценивается в результате определения плотности жировых и нежировых структур в неконтрастную фазу (жировые структуры имеют пониженную плотность). Богатые липидами ткани характерны для доброкачественных аденом коры надпочечника. Однако около 25% доброкачественных аденом могут не иметь низкой «неконтрастной» плотности. При исследовании КТ-плотности на разных фазах выведения контраста получены данные, что аденомы коры надпочечника быстро снижают показатели плотности (через 10 мин. после введения контраста, т.н. показатель “wash-out”, более чем на 50%), в то время как другие образования надпочечников имеют тенденцию к задержке контрастного вещества. Измерение этого показателя имеет близкую к абсолютным значениям диагностическую ценность при дифференциальном диагнозе аденом с одной стороны, от ФХЦ/ПГ, АКР и метастатической карциномы с другой.
Низкая (менее 10-15 HU) нативная плотность тканевого компонента при КТ или быстрое снижение интенсивности сигнала после внутривенного контрастирования абсолютно нехарактерны для АКР, метастазов и ФХЦ/ПГ.
Дифференциальный диагноз с ФХЦ/ПГ осуществляется на основании биохимического исследования норметанефрина и метанефрина.
МРТ и УЗИ обладают высокой чувствительностью в выявлении опухолей надпочечников, однако специфичность методов значительно ниже в связи с отсутствием четких количественных денситометрических показателей.
Дополнительным существенным преимуществом КТ является возможность оценки критерия плотности опухоли в фазу вымывания (wash-out).
Метастатическое поражение должно быть исключено/подтверждено в первую очередь у пациентов с анамнезом онкологического заболевания. Также, вероятность метастатического поражения рассматривается при двустороннем поражении надпочечников, особенно при отсутствии явлений гормональной активности, при наличии КТ-признаков, характерных для метастазов. Пациенты с подобными поражениями должны проходить онкологическое обследование для исключения распространенного опухолевого процесса (в первую очередь рака легкого, желудка, колоректального рака).
• 2.4.2. Рекомендуется пациентам с подозрением на опухоль надпочечника и наличием противопоказаний к выполнению КТ с контрастным усилением провести МРТ органов брюшной полости (ОБП) и забрюшинного пространства в рамках первичной диагностики [5,8].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 5
Комментарий. МРТ обладает высокой чувствительностью в выявлении опухолей надпочечников оценки распространенности процесса, в том числе поражения смежных структур.
Однако специфичность методов значительно ниже КТ в связи с отсутствием четких количественных денситометрических показателей [9].
• 2.4.3. Рекомендуется пациентам с опухолью надпочечника небольшого размера (до 4 см) выполнить позитронную эмиссионную томографию с 18-фтордезоксиглюкозой, совмещенную с компьютерной томографией (18ФДГ-ПЭТ/КТ), для верификации злокачественного потенциала. Высокая метаболическая активность (SUV более 3) является критерием, позволяющим с высокой степенью вероятности предполагать злокачественную природу опухоли [40-43].
Уровень убедительности рекомендаций - В, уровень достоверности доказательств - 2
Комментарий. В случае подозрения на АКР или метастазы при КТ-плотных образованиях малого размера (например, до 4 см) показано проведение 18ФДГ-ПЭТ/КТ, так как метод позволяет определить метаболическую активность образования. Если определяемый 18ФДГ-ПЭТ/КТ накопительный критерий SUV (standartised uptake value) более 3, то вероятность злокачественного поражения превышает 80% [39-43].
2.4.4. Рекомендуется пациентам с высоковероятным или клинически установленным диагнозом АКР выполнить спиральную компьютерную томография легких для оценки возможного метастатического поражения [5,8,13].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 5
Комментарий. Поскольку выбор оптимальной лечебной тактики и, в частности, вопросы хирургического лечения, определяются стадией заболевания, необходимо адекватно оценить распространенность опухолевого процесса. КТ ОГК, ОБП, забрюшинного пространства и малого таза с КУ позволяет выявить подавляющее большинство метастатических локаций, которые чаще всего включают легкие и печень.
Для метастазов брюшной полости есть преимущества и недостатки при использовании как КТ, так и МРТ, но для грудной полости КТ с КУ является предпочтительным методом, так как она превосходит все другие методы в обнаружении пульмональных метастазов малых размеров [9].
• 2.4.5. Рекомендуется пациентам с высоковероятным или клинически и/или патоморфологически верифицированным диагнозом АКР и подозрением на метастатическое поражение костей скелета выполнить сцинтиграфию костей всего тела или 18ФДГ-ПЭТ/КТ для уточнения наличия/отсутствия костных метастазов, а также провести МРТ головного мозга для исключения метастатического поражения головного мозга при наличии клинически обоснованных подозрений [5,8,13].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 5
Комментарий. Изолированные метастазы в кости и головной мозг являются редкими событиями при АКР без метастатических поражений других локализаций. Дополнительная визуализация, фокусирующаяся на этих участках, оправдана только тогда, когда имеется обоснованное клиническое подозрение или выявлены вторичные очаги иных локализаций. Следует отметить, однако, что этот вопрос в настоящее время изучен недостаточно и требуется проведения дальнейших исследований [9].
• 2.4.6. Выполнение пункционной биопсии опухоли надпочечника может быть рекомендовано только при обоснованном подозрении на его метастатическое (вторичное) поражение или в случае неоперабельного метастатического опухолевого процесса перед началом лекарственной противоопухолевой терапии. В остальных случаях выполнение пункционной биопсии опухоли надпочечника категорически не рекомендуется. [8,13,122].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 5
Комментарий. В дифференциальном диагнозе органоспецифичных опухолей надпочечника пункционная биопсия не имеет доказанных преимуществ, ассоциируется с низкой чувствительностью, специфичностью а также с вероятностью осложнений, в том числе метастатического прогрессирования. Тотальное выполнение пункционной биопсии опухолей надпочечников не продемонстрировало улучшения результатов дифференциальной диагностики инциденталом, напротив, привело к росту осложнений, ложноположительным и ложноотрицательным заключениям. В настоящий момент пункция целесообразна лишь при подозрении на метастатическое поражение надпочечников, где чувствительность цитологического исследования составляет 80-86%. Так же пункционная биопсия показана при подозрении на неходжкинскую лимфому с изолированным поражением надпочечников. Заболевание встречается крайне редко, характеризуется двусторонним инфильтративным поражением надпочечников, быстрым темпом роста опухоли, а также косвенными признаками, позволяющими подозревать лимфому, такими как выраженные явления общей интоксикации, повышение лактат-дегидрогеназы в сыворотке крови и т.д. При других вариантах надпочечниковых опухолей диагностическая ценность предоперационной пункционной биопсии неудовлетворительна (чувствительность не превышает 65%) [44].
В настоящий момент пункционная биопсия для диагностики ФХЦ/ПГ не рекомендована [45].
2.5 Иные диагностические исследования
2.5.1 Стадирование АКР
• 2.5.1.1. Рекомендуется всем пациентам с высоковероятным или верифицированным диагнозом АКР провести тщательное топическое обследование в отношении возможных метастазов, опухолевых тромбов и местного распространения опухоли, а также стадирование опухолевого процесса для определения лечебной тактики (см. также разд. 1.5.2) [8,13,46,47,45].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 5
Комментарий. Размер опухоли при АКР - один из важнейших клинических факторов прогноза, поэтому он является основой стадирования данного заболевания. Долгое время применялись авторские классификации АКР (MacFarlane, Icard, Lee). Критерии, определяющие символ «Т» по классификации MacFarlane, легли в основу классификации UICC и ENSAT. До последней редакции классификации TNM UICC опухоли T3-4N1 относились к IVстадии, а по классификации ENSAT -к III стадии, поэтому последняя считалась прогностически более точной.
В 2015г. проводили повторный анализ данных регистра ENSAT по АКР (включающий 463 пациента) относительно прогностической значимости [126]. Было показано, что венозная инвазия не ухудшает прогноз у пациентов с III стадией; поражение лимфоузлов было отнесено к TV стадии. При этом, как отмечают авторы, накоплено больше данных по венозной инвазии и результатам хирургического лечения этих пациентов, что нельзя говорить о поражении лимфоузлов при АКР.
80% взрослых пациентов на момент первичного выявления имеют размер опухоли не менее 10 см, у 30-40% пациентов предоперационно выявляются метастазы. Вероятность наличия отдаленных метастазов при опухолях более 10 см по данным послеоперационного наблюдения составляет более 80% [46,47, 48].
Определение распространенности опухолевого процесса крайне важно с точки зрения выбора лечебной тактики. В спектр исследований при поиске метастазов необходимо включение КТ легких, КТ или МРТ головного мозга и брюшной полости, сцинтиграфии и МРТ костей, 18ФДЕ-ПЭТ/КТ. Для выявления опухолевых тромбов выполняется флебография забрюишнных сосудов.
В случае АКР серьезное влияние на тактику может оказать степень гормональной активности опухоли. Так, если при гормонально-неактивной опухоли оперативное лечение распространенных форм АКР не имеет известных преимуществ, то при гормональноактивной опухоли, сопровождающейся тяжелыми соматическими проявлениями гиперкортицизма может быть оправдана тактика, направленная на максимальную циторедукцию.
2.5.2. Патолого-анатомическая диагностика
• 2.5.2.1. Рекомендуется всем пациентам с опухолью надпочечника выполнить патологоанатомическое исследование биопсийного (операционного) материала, включающее одну из принятых систем балльной оценки злокачественного потенциала опухоли, с применением иммуногистохимических методов с целью оценки органной принадлежности опухоли и ее злокачественного потенциала. При констатации злокачественного потенциала опухоли рекомендуется всем пациентам с опухолью надпочечника провести определение индекса пролиферативной активности экспрессии Ki-67 иммуногистохимическим методом для оценки прогноза течения заболевания [9,13,48-52,119-123].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 4
Комментарий. Существует несколько систем балльной оценки злокачественного потенциала опухоли надпочечников. В настоящее время наиболее распространена система Weiss, включающая 9 критериев световой микроскопии [48,49]:
• высокий ядерный индекс,
• количество митозов более 5 на 50 полей зрения при большом увеличении,
• патологические митозы,
• менее 25% клеток со светлой цитоплазмой,
• диффузный характер роста более 33% опухолевой ткани,
• некрозы,
• инвазия опухоли в венозное русло,
• инвазия опухоли в синусоиды,
• инвазия опухоли в капсулу.
Каждый из 9 критериев оценивается в 1 балл. При наборе 3 баллов и более по шкале Weiss подтверждается диагноз АКР.
При использовании критериев Weiss митотическую активность оптимально оценивать при большом увеличении по меныией мере в 10 различных полях зрения в пределах участка с наибольшим количеством митозов в каждом из 5 стекол. Минимальное количество оцениваемых полей зрения составляет 50.
Существует альтернативная по подсчету баллов, схожая по набору параметров световой микроскопии система van Slooten и соавт. [50] для дифференцировки доброкачественных и злокачественных адренокортикальных опухолей, панель включает в себя оценку следующих параметров:
• обширные регрессивные изменения (некроз, кровоизлияния, фиброз, кальцификация) -5,7 баллов,
• потеря нормальной структуры -1,6 баллов,
• ядерная атипия -2,1 баллов,
• ядерная гиперхромия -2,6 баллов,
• неправильные ядрышки -4,1 баллов,
• митотическая активность (>2 в 10 полях зрения при большом увеличении) -9,0 баллов,
• сосудистая или капсульная инвазия -3,3 балла.
Гистологический индекс >8 баллов по шкале van Slooten констатирует высокий злокачественный потенциал опухоли надпочечника.
Сравнительное изучение диагностической ценности балльной оценки перечисленных систем несколько отличается у разных исследователей, однако доказана несомненная польза их применения [51, 52], чувствительность шкал в отношении АКР и метастазов составляет около 92% и значительно возрастает при использовании консилиума патологов. На сегодняшний день в мире наиболее широко используется система Weiss [9].
По рекомендациям экспертов ВОЗ, при онкоцитарном морфологическом варианте адренокортикальной опухоли рационально использовать модифицированную систему балльной оценки злокачественного потенциала Lin-Weiss-Bisceglia [9,13].
При сомнительной органной принадлежности или неопределенном злокачественном потенциале опухоли ведущее значение приобретает иммуногистохимическое исследование. Наиболее часто с этой целью используют оценку интенсивности экспрессии белка р53, Ki67, кальретинина, цитокератинов 8 и 18, виментина, ингибина-а, мелана А, стероидогенного фактора-1 (SF-1) стероидного рецептора коактиватор-1 (SRC-1), протеина S-100, Рах8, хромогранина А, нейроспецифической энолазы, синаптофизина. В дополнение к диагностическим алгоритмам при дифференциальной диагностике адренокортикальной аденомы и АКР возможно использование иммуногистохимического маркера К167: для аденомы характерен индекс пролиферации Ki67 менее 5%, для АКР - более 5% [2, 13].
Предметом дискуссии до настоящего времени является прогностическое значение количества Weiss- или van Slooten-баллов, однако балльная система параметров световой микроскопии в качестве прогностического критерия уступает пролиферативному индексу Ki67. При повышении индекса Ki67 более 10% вероятность рецидива АКР после адекватной резекции в пределах здоровых тканей (RO-резекция) составляет не менее 80% (см. также
Приложение Г Табл. 18). Прогностическое значение других маркеров ИГХ в отношении АКР в настоящий момент находится в стадии исследования и накопления анализируемого материала.
• 2.5.2.2. Рекомендуется всем пациентам с опухолью надпочечника проводить патологоанатомическое исследование биопсийного (операционного) материала по протоколу, включающему, как минимуму, следующие данные: количество баллов по шкале Weiss или иной шкале балльной оценки злокачественного потенциала опухоли, пролиферативный индекс Ki67, резекционный статус (инвазия капсулы опухоли, и/или окружающих тканей, и/или прилежащих органов или ее отсутствие), наличие или отсутствие поражения лимфатических узлов [119-123].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 4
Комментарий. Важность оценки злокачественного потенциала опухоли по системе Weiss или иной шкале балльной оценки, а также пролиферативного индекса Ki67 обсуждены выше (см. разд. 2.5.2.1.). Необходимость оценки резекционного статуса опухоли (см. также Приложение Г Табл. 18), а также наличия или отсутствия поражения лимфатических узлов продиктована прогностической ценностью этих данных [9].
2.5.3 Генетическое тестирование
• 2.5.3.1. Рекомендуется всем пациентам с верифицированным диагнозом АКР провести, как минимум, стандартное генетическое консультирование, включающее изучение анамнеза жизни, анамнеза заболевания и семейного анамнеза, для выявления или исключения наследственного синдрома, ассоциированного с АКР [1,4].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 5
Комментарий. Своевременное выявление герминативной мутации и, соответственно, диагностика наследственного синдрома, включающего АКР, позволяют прогнозировать развитие других ассоциированных заболеваний у пациента, а также целенаправленно обследовать его кровных родственников.
Среди взрослой популяции до 5% случаев АКР развивается в рамках синдрома Ли-Фрумени, обусловленного герминативной мутацией в гене ТР53, около 3% - в рамках синдрома Линча. До 13% пациентов с синдромом МЭН 1 типа имеют АКР. АКР описан также как компонент синдромов Гарднера (семейный аденоматозный полипоз), комплекса Карни, Беквита-Вайдемана, нейрофиброматоза 1 типа (см. также разд. 1.2).
Выделяют ряд клинических особенностей, которые позволяют заподозрить наследственный характер АКР (Табл. 5) [4].
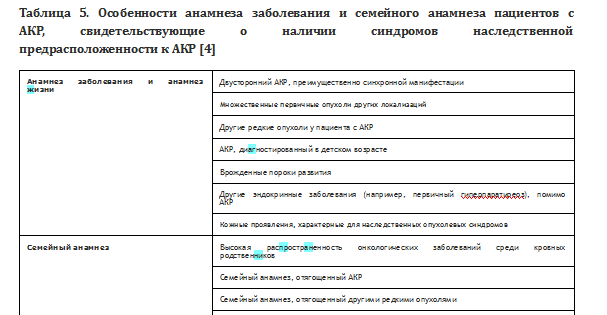
Анамнез заболевания и анамнез жизни Двусторонний АКР, преимущественно синхронной манифестации
Множественные первичные опухоли других локализаций
Другие редкие опухоли у пациента с АКР
АКР, диагностированный в детском возрасте
Врожденные пороки развития
Другие эндокринные заболевания (например, первичный гиперпаратиреоз), помимо АКР
Кожные проявления, характерные для наследственных опухолевых синдромов
Семейный анамнез Высокая распространенность онкологических заболеваний среди кровных родственников Семейный анамнез, отягощенный АКР
Семейный анамнез, отягощенный другими редкими опухолями Пациентам с АКР может быть рекомендован молекулярно-генетический скрининг на предмет выявления синдромов Ли-Фраумени и Линча, которые встречаются у в 3-5% случаев АКР и имеют достаточно разработанные клинические рекомендации.
Распространенность герминативных мутаций de novo в гене ТР53 достигает 20-25% среди всех пациентов с аберрациями в данном гене, очевидно, что такие пациенты не будут иметь АКР или иных проявлений синдрома Ли-Фраумени в семейном анамнезе. В связи с этим эксперты ВОЗ, Европейского общества эндокринологов (European Society of Endocrinology) и ENSAT рекомендуют скринировать всех пациентов с АКР на наличие герминативных мутаций в гене ТР53.
Синдром Линча присутствует у той же части пациентов с АСС, что и у пациентов с колоректальным раком (3-5%), которым показан скрининг на синдром Линча. Его диагностика может проводиться путем непосредственного молекулярно-генетического исследования генов MSH2, MLH1, PMS2, MSH6 и ЕРСАМ или путем иммуногистохимического исследования с антителами к MSH2, MLH1, PMS2, MSH6 и оценки микросателлитной нестабильности.
50-90% случаев АКР в детском возрасте наблюдается в рамках синдрома Ли-Фраумени. В связи с этим в случае выявления АКР в детском возрасте, всем пациентам необходимо проведение молекулярно-генетического анализа для выявления патогенных герминативных мутаций в гене ТР53 [4, 2, 9].
Пациентам с АКР может быть рекомендован молекулярно-генетический скрининг на предмет выявления синдромов Ли-Фраумени и Линча, которые встречаются у в 3-5% случаев АКР и имеют достаточно разработанные клинические рекомендации.
Распространенность герминативных мутаций de novo в гене ТР53 достигает 20-25% среди всех пациентов с аберрациями в данном гене, очевидно, что такие пациенты не будут иметь АКР или иных проявлений синдрома Ли-Фраумени в семейном анамнезе. В связи с этим эксперты ВОЗ, Европейского общества эндокринологов (European Society of Endocrinology) и ENSAT рекомендуют скринировать всех пациентов с АКР на наличие герминативных мутаций в гене ТР53.
Синдром Линча присутствует у той же части пациентов с АСС, что и у пациентов с колоректальным раком (3-5%), которым показан скрининг на синдром Линча. Его диагностика может проводиться путем непосредственного молекулярно-генетического исследования генов MSH2, MLH1, PMS2, MSH6 и ЕРСАМ или путем иммуногистохимического исследования с антителами к MSH2, MLH1, PMS2, MSH6 и оценки микросателлитной нестабильности.
50-90% случаев АКР в детском возрасте наблюдается в рамках синдрома Ли-Фраумени. В связи с этим в случае выявления АКР в детском возрасте, всем пациентам необходимо проведение молекулярно-генетического анализа для выявления патогенных герминативных мутаций в гене ТР53 [4, 2, 9].
нарушения кровотока по ней и развития коллатерального кровотока, на основании предоперационной оценки;
• высокая вероятность эмболических осложнений является основной причиной (!) интраоперационной и ранней послеоперационной смертности у пациентов с АКР. Эмболия может быть обусловлена несколькими факторами или их сочетанием:
• наличием гиперкортицизма/гиперандрогении, способствующих исходной гиперкоагуляции;
• массивной интраоперационной кровопотерей с одномоментным переливанием больших объемов компонентов крови;
• наличием опухолевых тромбов;
• отсутствием предоперационных профилактических мероприятий, снижающих риск тромбоэмболии.
• Среди профилактических мероприятий должны рассматриваться гепаринопрофилактика, установка венозных тромбоулавливающих зондов. Последнее может быть затруднено из-за опухолевой компрессии нижней полой вены.
Нарушение капсулы опухоли практически в 100% наблюдений приводит к местным рецидивам АКР. Прецизионность манипуляций при удалении опухоли является одной из ключевых задач хирурга, так как нарушение целостности капсулы приводит к местной опухолевой диссеминации даже после выполнения RO-резекции [8, 53, 54].
При АКР показано удаление регионарных лимфоузлов в случае их метастатического поражения или при подозрении на поражение по клинико-инструментальным данным [44,55].
Преимущества лимфодиссекции при АКР продемонстрированы в исследовании J. Reibetanz и соавт. [56], где на достаточно большой выборке пациентов доказано статистически значимое улучшение безрецидивной выживаемости и АКР-ассоциированной смертности у пациентов с RO-резекцией. В качестве дополнительного довода приводится факт, что в группе с лимфодиссекцией средний размер опухоли был достоверно больше и мулыпивисцеральная резекция выполнялась чаще, однако, несмотря на это, достигнуты лучшие результаты. В исследовании Gaujoux S. и соавт. [57] указывается, что вовлеченность лимфоузлов в опухолевый процесс у пациентов с АКР, неранжированных по стадиям, составляет 20%, что является, по мнению авторов, показанием к более широкому выполнению лимфодиссекции. Границы превентивной лимфодиссекции не точно определены, по имеющимся данным подлежат удалению лимфоузлы ворот почки, паракавальные справа и парааортальные слева.
При отсутствии явных признаков инвазии в паренхиму или ворота почки, необходимости в “превентивной" нефрэктомии (нефрадреналэктомии), для повышения радикальности резекции, нет. Достаточна резекция жировой капсулы почки с отступлением от краев опухоли на 2-Зсм.
• 3.1.3. Пациентам с клинически верифицированным диагнозом АКР может рекомендоваться выполнение эндоскопической (лапароскопической, ретроперитонеоскопической) адреналэктомии при сочетании следующих факторов: размер опухоли менее 6 см, I и II стадия заболевания по ENSAT (отсутствие признаков инвазии опухоли в окружающие структуры), достаточный опыт подобных вмешательств у хирурга и лечебного учреждения. При большей распространенности процесса эндоскопическая операция противопоказана, так как возможность выполнения лимфаденэктомии и адекватной ревизии и резекции соседних органов ограничена, радикальность вмешательства сомнительна [8,13,59,66].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 4
Комментарий. Основная проблема лапароскопической адреналэктомии - высокая частота диссеминации опухоли по брюшине после операции, вследствие интраоперационного повреждения капсулы опухоли. Часть экспертов придерживается мнения, что при размере опухоли более 5 см и/или подозрении на злокачественность опухоли по результатам предоперационного обследования - вероятность диссеминации опухоли при нарушении капсулы значительно выше, чем при открытом вмешательстве [8,44,47, 53, 58 - 60].
Недискутабельным пунктом данной рекомендации является специализированный характер учреждения и опыт операционной бригады. •
• 3.1.4. Рекомендуется пациентам с резектабельными формах местного рецидива или при солитарных/единичных метастазах, возникших после операции, выполнить повторную RO-резекцию как наиболее эффективный и предпочтительный вариант лечения, обеспечивающий наиболее длительный безрецидивный период или продолжительность жизни (см. также Приложение Б Рис.1). Безрецидивный период более 1 года после первой операции является благоприятным прогностическим фактором при хирургическом лечении рецидивов или метастазов АКР. Целесообразность повторных операций при рецидивах определяется индивидуально, муьтидисциплинарным консилиумом, с учетом распространённости процесса, вероятности полной циторедукции, биологических особенностей опухоли и возможности консервативного лечения [58,62,64,65].
Уровень убедительности рекомендаций - В, уровень достоверности доказательств - 3
Комментарий. Частота прогрессирования АКР после хирургического лечения в течение 5 лет достигает до 80-85%. Единственным методом, обеспечивающим длительное выживание при рецидивах или метастазах остается повторная «радикальная», то есть R0- резекция. Ее возможность следует рассматривать у каждого пациента с учетом распространенности процесса, особенностей предыдущего хирургического лечения, биологических особенностей опухоли, таких как степень злокачественности и темп роста.
В онкологическом центре SIoan-Kettering, США, повторные резекции выполнены у 47 пациентов с местным рецидивом или отдаленными метастазами АКР [61]. При этом из ИЗ пациентов в 62 случаях операция была радикальной, в 45 - паллиативной. Среди радикальных операций 43 (69%) выполнено по поводу отдаленных метастазов, 14 (23%) - по поводу местного рецидива, 5 (8%) - по поводу и рецидива и метастазов. Все операции по поводу местного рецидива были комбинированные, с резекцией пораженных смежных органов, среди локализаций отдаленных метастазов самыми частыми были печень (28), легкие (17), брюшина (8). Медиана общей выживаемости пациентов подвергшихся повторной радикальной резекции составила 74 мес, 5-летняя выживаемость - 57%; у пациентов после паллиативной повторной резекции - 16 мес и 0% соответственно, различия были статистически значимы. Прогноз пациентов, оперированных по поводу местного рецидива или отдаленного метастазов, не отличался.
По данным французской ассоциации эндокринных хирургов [62] повторные операции выполнены у 22 пациентов. 5-летняя выживаемость с момента установки диагноза составила 27%, с момента повторной операции -16% (при радикальной повторной резекции - 28%).
R. Bellantone и соавт. [63] в 1997 г анализировали данные Итальянского регистра по АКР, включающего 188 пациентов. Средняя продолжительность жизни пациентов оперированных повторно по поводу рецидива составила 15,9 мес., против 3,9 мес. у пациентов, получавших только консервативное лечение, 3-летняя выживаемость составила 23% и 0% соответственно.
В Туринском университете [64] ретроспективно анализировали сравнительный опыт лечения рецидивов АКР: группа А - 22 пациентов оперированы, 17 - в объеме удаления рецидива, 5 -удаление отдаленного метастаза (средний балл Weiss - 6, среднее время до прогрессирования -22 мес, средний Ki67 - 18%). Еруппа В - 17 пациентов получили химиотерапию по схеме #доксорубицин• ** этопозид** Щисплатин** и митотан** (средний балл Weiss - 7, среднее время до прогрессирования - 9,5 мес, средний Ki67 - 28%). В группе А у 5 пациентов (22%) рецидива не наблюдалось, у 17 возник рецидив в среднем через 23 мес, 8 пациентов повторно оперированы; средняя общая выживаемость составила 86 мес. В группе В 88% пациентов за время наблюдения умерли от прогрессирования; средняя общая выживаемость составила 33,5 мес -достоверно меньше.
Тем не менее, во всех приведенных исследованиях нельзя исключить неравнозначность распределения пациентов, вследствие того, что повторно не оперировали пациентов с большим распространением процесса или с заведомо плохим прогнозом. Индивидуализация показаний к повторным операциям является нерешенной задачей и требует дальнейших исследований с учетом также биологических особенностей опухоли [65].
• 3.1.5. Всем пациентам с АКР и гиперкортицизмом (манифестным или субклиническим) рекомендуется проведение заместительной терапии надпочечниковой недостаточности в послеоперационном периоде [13,19,24].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 5
Комментарий. Манифестный гиперкортицизм и функционально автономная гиперпродукция кортизола, как правило, приводят к развитию надпочечниковой недостаточности после адреналэктомии с опухолью. В связи с этим всем пациентам с АКР и верифицированным/ высоковероятным гиперкортицизмом показано проведение заместительной терапии надпочечниковой недостаточности интраоперационно и в послеоперационном периоде в соответствии с клиническими рекомендациями [9].
3.2. Консервативное лечение
• 3.2.1. Не рекомендуется проведение адъювантной терапии пациентам с адренокортикальной опухолью неопределенного злокачественного потенциала [73,85,86,95].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 5
Комментарий. Адъювантные методы лечения связаны с потенциальной токсичностью, в связи с этим адъювантную терапию следует рассматривать только для пациентов с верифицированным патоморфологически диагнозом АКР (см также разд. 2.5.2.1) [9].
• 3.2.2. Рекомендуется всем пациентам после выполнения RO-резекции по поводу АКР при Ki67 более 10% проводить адъювантное лечение митотаном** (орто-пара-DDD) с целью эрадикации или длительного подавления микрометастазов [73,85,86,95].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 4
Комментарий. Терапия митотаном** должна быть начата в кратчайшие сроки после хирургического лечения. Уровень препарата в крови требует постоянного лабораторного мониторирования и, при условии его переносимости, рекомендованный диапазон терапевтической концентрации составляет от 14 до 20 мкг/мл. Доза митотана** необходимая для достижения и/или поддержания его концентрации в крови в терапевтическом диапазоне, индивидуальна, как правило, составляет 4-8 г/сут. При отсутствии рецидива АКР адъювантное лечение проводится от 2 до 5 лет (см. также Приложение АЗ).
В 2004 г. по решению EMA (European Medicine Agency) в качестве базового препарата для лечения АКР предложен 1 - (2-хлорфенил) -1 - (4-хлорфенил) -2,2-дихлорэтан. Это единственный, в настоящее время безальтернативный по эффективности препарат, специфично действующий на пучковую, частично сетчатую зону, и приводящий к клеточной дегенерации коры надпочечника, при этом, не затрагивая клубочковую зону. Наряду с противоопухолевым воздействием, митотан** обладает адренолитическим эффектом, повреждая внутриклеточные ферменты, участвующие в синтезе стероидов, таким образом, снижая интенсивность надпочечникового стероидогенеза [66, 67]. В связи с блокированием стероидогенеза пациенты, получающие митотан** нуждаются в заместительной терапии глюкокортикоидами. Уменьшение уровня глюкокортикоидов в крови связано не только с адренолитической активностью митотана**, но и с индукцией им печеночного клиренса стероидов [68], в связи с чем необходимо минимум двукратное увеличение стандартной дозы заместительной терапии надпочечниковой недостаточности.
Недостаточная доза заместительной терапии снижает переносимость митотана** и утяжеляет его побочные эффекты.
Препарат используется в качестве основного средства химиотерапии АКР с 1970 года, рекомендован FDA - Agency of Food and Drug Administration, USA). В феврале 2018г. митотан** впервые зарегистрирован в России и разрешен к применению. Препарат включен в реестр жизненно необходимых и важнейших лекарственных препаратов (ЖНВЛП) [69].
Исследования по эффективности митотана** в адъювантном режиме, проводимые до 2006 года, отличались противоречивыми выводами. Если адъювантная терапия проводилась при размерах первичной опухоли более 10 см, то при сравнении с контрольными группами пациентов, не получавших препарат, но с меньшими размерами опухолей - получены парадоксальные результаты, свидетельствующие о лучших лечебных показателях в контрольных группах пациентов, не получавших митотан** [70, 71].
В мулыпицентровом исследовании и мета-анализе немецко-итальянского регистра пациентов с АКР [72] анализированы результаты лечения 177 пациентов и продемонстрировано увеличение безрецидивного периода при проведении адьювантной терапии -42мес против 10 и 25 мес в двух контрольных группах и снижение частоты рецидива до 49% против 73% и 91% соответственно.
При анализе эффективности митотана** у 246 пациентов с распространенными формами АКР (IV стадия ENSAT) в 26% наблюдений отмечен объективный ответ в соответствии с критериями RECIST (И пациентов - полный ответ, 52 - частичный ответ) [75]. Нужно отметить, что в большинстве исследований по эффективности митотана** при распространенных формах АКР отмечаются следующие общие тенденции и выводы:
• случаи полного излечения имеют место, но являются крайней редкостью;
• частичный или полный опухолевый ответ в подавляющем большинстве отмечается у пациентов с достижением терапевтической концентрации митотана** в крови [76-79].
• независимо от достижения целевой терапевтической концентрации отмечается увеличение общей выживаемости, более длительной при целевых значениях митотана** в крови [70-74,80-82].
• на фоне приема митотана** в большинстве наблюдений удается контролировать проявления гиперкортицизма [8].
• достижение целевой концентрации митотана** в крови не всегда зависит от принимаемой дозы препарата, в большей степени зависит от кумуляции дозы, связанной с длительным периодом полужизни препарата, что доказывает необходимость тщательного мониторинга уровня митотана** в крови и постоянной коррекции дозы [78-79].
• митотан** имеет узкий терапевтический диапазон дозирования, ограниченный, с одной стороны, необходимостью быстрейшего достижения терапевтической концентрации препарата более 14 мг/л, с другой - переносимостью препарата. В целевых концентрациях частота побочных эффектов составляет не менее 80% наблюдений (минимум 1 побочный эффект), интенсивность и амплитуда нежелательных клинических проявлений нарастает с проявлениями кумуляции дозы, проявления исчезают с отменой препарата (Табл. 6) [8].
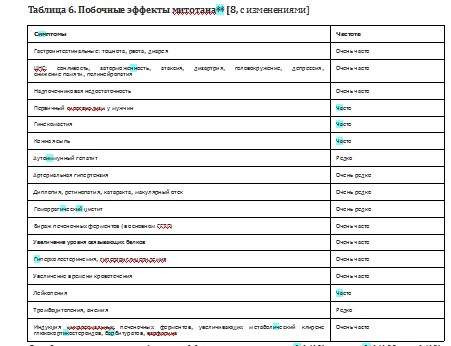
Определение частоты побочных эффектов: очень часто (>1/10), часто (>1/100 to <1/10), нечасто (>1/1000 to <1/100), редко (от >1/10 000 до <1/1000), очень редко гаге (<1/10 000), не известно (не может быть определено на основании имеющихся на сегодняшний день результатов исследований).
Таким образом, митотан** показал значительную эффективность в отношении безрецидивной выживаемости после RO-резекций и улучшение показателей общей выживаемости при распространенных формах АКР.
• 3.2.3. Рекомендуется всем пациентам, которым показана терапия митотаном**, начинать лечение с небольших доз с постепенной эскалацией, под контролем состояния пациента, переносимости лечения, концентрации митотана** в плазме крови (см. также Приложение АЗ) [83-85].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 4
Комментарий. Существуют разные схемы терапии митотаном** ни одна из них не обладает доказанным преимуществом на сегодняшний день. Наиболее широко используется схема, предполагающая старт терапии с дозы 1 г/сут с постепенной эскалацией дозы на 0.5-1 г/сут каждые 3-7 дней до суммарной дозы в 4 г/сут. При хорошей переносимости митотана** может быть использован подход с высокой начальной дозой 1.5 г/сут и быстрым ее приращением до 6 г/сут. Дальнейшая коррекция дозы митотана** проводится в соответствии с его концентрацией в крови (необходимо определение через 4 недели от момента выхода на дозу 4 или 6 г/сут) и переносимостью [83-85]. В режиме высоких доз необходимо контролировать уровень митотана** в крови через 2-3 недели после начала терапии.
В сравнительном фармакокинетическом исследовании режим с высокими дозами на старте терапии начальный позволил достичь терапевического диапазона выше 14 мкг/мл у большего количества пациентов. Однако эти результаты не были статистически значимыми из-за недостаточной мощности исследования. Помимо этих двух режимов, существует множество других вариаций; выбор зависит от опыта врача, особенностей клинического течения АКР и состояния пациента. Рекомендации по применению митотана** в качестве адьювантной терапии см. также в Приложении АЗ.
Митотан** является липофильным лекарственным средством и, как предполагается, лучше всасывается из кишечника при совместном потреблении с жиросодержащими продуктами, например, с молоком или шоколадом. В случае ограниченной желудочно-кишечной толерантности должно быть предложено симптоматическое лечение тоитоты/рвоты и/или диареи.
У всех пациентов, получающих терапию митотаном** необходимо проверять его лекарственные взаимодействия с другими препаратами (в частности, из-за индукции цитохрома CYP3A4 под влиянием митотана**).
Все сопутствующие лекарства должны быть проверены на предмет метаболизма CYP3A4 и заменены альтернативными, если это возможно.
Молекулярные механизмы эффектов митотана** до сих пор изучены недостаточно. Тем не менее, известно, что митотан** повышает экспрессию печеночной монооксигеназы CYP3A4, метаболизирующей ряд лекарственных веществ. В связи с этим, все препараты, которые назначаются пациенту одновременно с приемом митотана** и/или на фоне присутствия его концентрации в крови, должны быть проверены на предмет метаболизма CYP3A4 и заменены альтернативными, если это возможно [86].
• 3.2.4. Рекомендуется принимать индивидуальное решение о возможности проведения адъювантной терапии АКР у пациентов с низким/промежуточным риском рецидива/ прогрессирования (см. также Приложение Б Рис.1) [95].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 4
Комментарий. На сегодняшний день, пролиферативный индекс Ki67 считается одним из ключевых предикторов рецидива АКР; пациенты с Ki67 <10% могут представлять собой подгруппу пациентов с относительно более благоприятным прогнозом. В связи с этим у пациентов с низким/промежуточным риском рецидива/прогрессирования (I-II стадии по ENSAT, RO-резекционный статус и Ki67 <>10%) адъювантная терапия митотаном** может быть чрезмерной, с учетом возможных нежелательных лекарственных реакций. На сегодняшний день не валидизировано ни одного клинического, гистопатологического или молекулярного маркера, который бы достоверно предсказывал реакцию на митотан**[9].
В настоящее время продолжается проспективное исследование ADJUVO в отношении пациентов с индексом Ki67 <10%. С учетом отсутствия достоверных данных решение о возможности проведения адъювантной терапии митотаном** у пациентов с низким/ промежуточным риском рецидива/прогрессирования АКР может быть принято в индивидуальном порядке при наличии факторов неблагоприятного прогноза.
• 3.2.5. Рекомендуется пациентам с неоперабельным АКР, наличием распространённого метастатического процесса после резекции первичной опухоли, быстрым прогрессированием заболевания проводить цитотоксическую терапию по схеме этопозид**, #доксорубицин**, #цисплатин** (режим приведен в таблице 7) - на фоне продолжающегося приема митотана** ежедневно в дозе, обеспечивающей его содержание в сыворотке крови на уровне 14-20 мкг/лм без перерыва между циклами химиотерапии (схема EDP-M) [93].
Уровень убедительности рекомендаций - А, уровень достоверности доказательств - 2
• Рекомендуется пациентам с невысокой опухолевой нагрузкой и медленной прогрессией рассмотреть вопрос о монотерапии митотаном** в комбинации с возможными местными циторедуктивными или радиологическими методами в качестве первичного лечения (см. также Приложение Б Рис.2) [8,13,93,95].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 4
Комментарий. Пациенты с неоперабельным АКР, наличием распространённого метастатического процесса после резекции первичной опухоли, быстрым прогрессированием заболевания должны получать цитотоксическую химиотерапию (см. также Приложение Г1 Табл. 15-17).
В течение длительного времени считалось, что АКР является стойким к стандартной цитостатической химиотерапии. [87-91].
Среди взрослых пациентов наиболее эффективным , по данным рандомизированного исследования FIRM-ACT (FIRM-ACT ClinicalTrials.gov number, NCT00094497), признана комбинация этопозида** #доксорубицина** и Щисплатина**, на фоне постоянного приема митотана** (схема EDP-M). Исследование включало 304 пациента с распространенным вариантом АКР. При сравнении частоты ответа на терапию EDP-M в сравнении со схемой стрептозотоцин+митотан** (S+M) показана достоверно более высокая частота ответа на EDP-M: 23,2% против 9,2% на S+M (Р<0,001), и большее время стабилизации заболевания (5мес против 2), но не показало значимых различий в общей выживаемости (14,8 мес против 12,0) [93].
Терапевтические опции для детской популяции пациентов с АКР также весьма ограничены и в целом совпадают с таковыми для взрослых [132] Доказательная база основана преимущественно на результатах исследований с участием взрослых пациентов; исследования с участием детей единичные, характеризуются малым объемом выборки. В исследовании с участием 11 детей с распространенным АКР полный или частичный ответ достигнут в 81% случаев (9 пациентов) при применении протокола EDP-M на фоне достижения терапевтической концентрации митотана** в крови [133]. Протокол EDP-M используется в качестве 1 линии полихимиотерапии, вне зависимости от возраста пациента (см. Табл. 7). В клинической практике для взрослых пациентов с АКР наиболее часто используется первый режим дозирования, в детской популяции - второй.

Курс химиотерапии повторяется каждые 28 дней
• 3.2.6. Рекомендуется пациентам с распространенным АКР в качестве терапии 2 линии комбинация #гемцитабина** и фторпиримидинов (#фторурацил** или #капецитабин**) -на фоне продолжающегося приема митотана** ежедневно в дозе, обеспечивающей его содержание в сыворотке крови на уровне 14-20 мкг/лм без перерыва между циклами химиотерапии [94].
Уровень убедительности рекомендаций - В, уровень достоверности доказательств - 3
Комментарий. Режимы химиотерапии, применяемые во 2 линии лечения распространенного рака коры надпочечников представлены в Табл. 8 [94]

• 3.2.7. Рекомендуется принять индивидуальное решение о возможности проведения полихимотерапии АКР в адъювантном режиме у пациентов с высоким риском рецидива/ прогрессирования (см. также Приложение Б Рис.1) [8,13].Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 5
Комментарий. Рутинное проведение цитотоксической полихимотерапии не целесообразно, с учетом рисков нежелательных лекарственных реакций.
На сегодняшний день, данные об использовании цитотоксических препаратов в адъювантном режиме ограничены. Тем не менее, большинство экспертов Европейского общества эндокринологов (European Society of Endocrinology) и ENSAT одобряют индивидуальное решение вопроса о возможности проведения полихимотерапии АКР в адъювантном режиме у пациентов с высоким риском рецидива/прогрессирования (например, Ki67> 30%, опухолевый тромб в нижней полой вене или Rl-резекция).
Пациенты с Р2-резекционным статусом должны рассматриваться как пациенты с распространенным АКР (см разд. 5.3.1 и Приложение Б Рис. 2).
3.3. Иное лечение
3.3.1. Дистанционная лучевая терапия
• 3.3.1.1. Дистанционная лучевая терапия (ДЛТ) является методом выбора для паллиативной терапии при метастазах АКР в кости и центральную нервную систему.
Рекомендуется пациентам с костными метастазами АКР решить вопрос о проведении паллиативной лучевой терапии с целью уменьшения болевого синдрома. Лучевая терапия может проводиться различными режимами фракционирования - 24Ер за 6 фракций за 2 недели, ЗОЕр за 10 фракций за 2 недели, 8Ер за 1 фракцию [8].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 5
Комментарий. Длительное время АКР считался радионечувствительной опухолью. В настоящее время известно, что ДЛТ является методом выбора для паллиативной терапии при метастазах АКР в кости и центральную нервную систему (30-40 Ерей). Исследуются возможности конформной лучевой терапии с ЗО-планировкой как при нерезектабельных формах заболевания, так и в качестве дополнительного метода местного воздействия после нерадикального хирургического лечения.
• 3.3.1.2. ДЛТ может применяться в дополнение к терапии митотаном** на индивидуальной основе у пациентов после R1 резекции. Рекомендуется пациентам с локализованной стадией адренокортикального рака после выполнения хирургического вмешательства при наличии признаков высокого риска локального рецидива (положительные края резекции) решить вопрос о проведении дистанционной 3D -конформной лучевой терапии на ложе удалённой опухоли в дополнение к терапии митотаном**. 3D-конформная лучевая терапия на ложе опухоли проводится РОД 2Ер до СОД 50-60Ер 5 раз в неделю [95-99].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 4
• Рекомендуется пациентам с прогрессирующим или рецидивирующим течением АКР в дополнение к хирургическим вмешательствам решить вопрос о проведении локальной лучевой терапии на область опухоли (в зависимости от прогностических факторов, локализации опухоли, предпочтений пациента) [95-99].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 4
Комментарий. По данным литературы, применение ДЛТ способно предотвратить местное рецидивирование, но не оказывает значительного влияния на риск отдаленного метастазирования и общую выживаемость [95-97].
Известно, что именно отдаленные метастазы являются причиной 40-60% рецидивов АКР и оказывают значительное влияние на прогноз пациента, и их труднее эффективно лечить. Тем не менее, профилактика осложнений, вызванных местными рецидивами, свидетельствует в пользу лучевой терапии. Адъювантная ДЛТ может быть особенно целесообразной у пациентов с резекцией R1.
Комбинация лучевой терапии и митотана** биологически обоснована [98, 99] и возможна, но может быть связана с большей токсичностью. Недопустима отсрочка терапии митотаном** и/или системной химиотерапии из-за проведения ДЛТ.
Высокий риск интраоперационнной опухолевой диссеминации не является показанием к проведению ДЛТ.
Длительное время АКР считался радионечувствительной опухолью. По данным литературы, применение ДЛТ способно предотвратить местное рецидивирование, но не оказывает значительного влияния на риск отдаленного метастазирования и общую выживаемость [95-97].
Комбинация лучевой терапии и митотана** биологически обоснована [98, 99] и возможна, но может быть связана с большей токсичностью. Недопустима отсрочка терапии митотаном** и/или системной химиотерапии из-за проведения ДЛТ.
Высокий риск интраоперационнной опухолевой диссеминации не является показанием к проведению ДЛТ.
3.3.2. Заместительная терапия надпочечниковой недостаточности при лечении митотаном**
3.3.2.1. Рекомендуется всем пациентам, получающим терапию митотаном**, проводить заместительную терапию надпочечниковой недостаточности. В случае гиперкортицизма необходимость проведения заместительной терапии должна обсуждаться индивидуально. Оптимальными препаратами являются гидрокортизон**. Прием митотана** приводит к индукции печеночного клиренса стероидов и повышению уровня кортизол-связывающего глобулина в крови, в связи с этим необходимо минимум двукратное увеличение стандартной дозы заместительной терапии надпочечниковой недостаточности [83-85, 93, 131].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 4
Комментарий. Заместительную терапию надпочечниковой недостаточности, обусловленной приемом митотана** как правило, начинают с первого дня его применения. Наиболее распространенной стратегией является старт терапии с использованием полной заместительной дозы глюкокортикоидов, которая должна, как минимум, в два раза превышать стандартную (см. также разд. 3.2.2). Альтернативная стратегия состоит в том, чтобы начать заместительную терапию в редуцированной дозе - 20 мг/сут гидрокортизона** с постепенной эскалацией в течение нескольких недель. Суммарная доза гидрокортизона** обычно составляет 50 мг/сут, однако некоторым пациентам требуются суточные дозы до 100 мг.
Не существует надежного лабораторного маркера для определения оптимальной дозы гидрокортизона. Адекватность заместительной терапии надпочечниковой недостаточности на фоне терапии митотаном** должна основываться на параметрах клинической картины, таких как артериальное давление, пульс, аппетит, тошнота/рвота, динамика массы тела. В качестве вспомогательных биомаркеров, помимо клинической картины, могут учитываться концентрации АКТГ и калия крови.
При присоединении интеркуррентных заболеваний, стрессе (психологические или тяжелые физические нагрузки) необходимо увеличивать дозу глюкокортикоидов в 1.5-3 раза на время острой фазы заболевания или момент воздействия стресса. В дальнейшем целесообразен возврат к обычной заместительной дозе, которую пациент принимал до болезни. В случае невозможности перорального приема глюкокортикоидов по любым причинам, возникновения любых острых состояний необходим переход на парентеральный путь их введения. Оптимальным препаратом также является гидрокортизон **.
У некоторых пациентов, получающих митотан**, могут наблюдаться симптомы минералкортикоидной недостаточности (гиперкалиемия, гипонатриемия, гипотония, слабость), несмотря на применение глюкокортикоидов в полной заместительной дозе. В этом случае необходимо решить вопрос о добавлении к терапии флудрокортизон**а, с учетом клинической картины, уровня электролитов и ренина/активности ренина плазмы. [9].
3.3.3. Антирезорбтивная терапия
• 3.3.3.1. Рекомендуется пациентам с АКР и костными метастазами проводить антирезорбтивную терапию [8,13].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 5
Комментарий. Метастазы в кости у пациентов с АКР ассоциированы с низким качеством жизни из-за болевого синдрома, в также с возникновением патологических переломов, компрессии спинного мозга и гиперкальциемии. В ходе нескольких рандомизированных исследований III фазы было показано, что что ингибиторы костной резорбции, такие как бисфосфонаты и деносумаб**, эффективны у пациентов с метастазами в кости при первичных злокачественных новообразований молочной железы, предстательной железы, легких. Достаточных данных для пациентов с АКР в настоящее время нет. Однако, основываясь на этих результатах, лечение пациентов с костными метастазами любых раков с помощью антирезорбтивной терапии стало общепринятой практикой. Поэтому у пациентов с АКР и костными метастазами целесообразно назначать бисфосфонаты в лечебных дозах в сочетании с потреблением кальция и витамина D.
У пациентов с АКР и гиперкортицизмом отмечен повышенный риск глюкортикоидного остеопороза и остеопоротических переломов. Этой группе пациентов целесообразно проведение антирезорбтивной терапии с использованием «антиостеопорозных доз» бисфосфонатов или деносумаба**. Поскольку риск переломов быстро снижается после нормализации уровня кортизола в крови, продолжение антиостеопоротической терапии обычно не требуется после купирования гиперкортицизма.
3.3.4. Контрацепция
• 3.3.4.1. Рекомендуется пациенткам репродуктивного возраста, получающим цитотоксическую химиотерапию и/или терапию митотаном** подобрать эффективные методы контрацепции [100-101].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 4
Комментарий. Цитотоксическая химиотерапии и терапия митотаном** ассоциированы с потенциальными тератогенными эффектами, в связи с чем наступление беременности на фоне применения этих методов лечения противопоказано. Для предотвращения наступления нежелательной беременности пациенткам репродуктивного возраста целесообразно применение эффективных методов контрацепции (см также разд. 5.4).
На сегодняшний день известно, что клетки АКР могут экспрессировать эстрогеновые рецепторы. Кроме того, в доклинических исследованиях было показано, что эстрогены могут стимулировать прогрессирование АКР за счет перекрестных взаимодействий с сигнальными путями инсулиноподобного фактора роста (IGF, insulin-like growth factor). В связи с этим предпочтение следует отдавать не содержащим эстрогены контрацептивным препаратам и барьерным методам контрацепции [100,101].
Диетотерапия и обезболивание не применяются.
4. Реабилитация
• 4.1. Рекомендуется проводить реабилитацию пациентов с АКР, ориентируясь на общие принципы реабилитации пациентов после проведенных хирургических вмешательств и/ или химиотерапии. Реабилитация пациентов, оперированных по поводу гормональноактивной опухоли, должна включать профилактику надпочечниковой недостаточности в послеоперационном периоде [13,19, 24].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 5
5. Профилактика
5.1. Профилактика АКР
Первичная профилактика АКР отсутствуют.
Вторичная профилактика прогрессирования АКР заключается в надлежащем лечении и регулярном обследовании пациентов в полном объеме (см. разд. 3 и 5.2).
5.2. Послеоперационное наблюдение пациентов с АКР
5.2.1. Всем пациентам с АКР рекомендуется регулярное обследование, включающее (1) визуализирующие исследования органов брюшной полости, малого таза, грудной клетки, (2) лабораторные исследования гормональной опухолевой секреции. В течение первых 2-х лет от момента постановки диагноза после радикального хирургического лечения обследование проводят не реже 1 раза в 2-3 месяца, далее - не реже 1 раза в 3 - 6 месяцев в течение последующих Зх лет. После 5-ти лет безрецидивного течения АКР интервалы могут быть увеличены до 6-12 месяцев. В случае распространенного/прогрессирующего АКР решение об оптимальной периодичности обследования принимается индивидуально; как правило, оно проводится не реже 1 раза в 2-3 месяца [1,8,9] (см также Приложение Б Рис 1. И Рис. 2).
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 5
Комментарий. Регулярное стадирование опухолевого процесса в динамике необходимо для своевременной коррекции лечебной тактики и прогнозирования течения заболевания.
Определение распространенности опухолевого процесса - выявления локо-регионарного рецидива или отдаленного метастазирования - проводится на основании КТ легких, КТ или МРТ головного мозга и брюшной полости, сцинтиграфии и МРТ костей, 18ФДГ-ПЭТ/КТ. До настоящего времени нет опубликованных исследований, посвященных оптимальной периодичности обследования при АКР. В связи с этим рекомендации основаны на мнении экспертов Европейского общества эндокринологов (European Society of Endocrinology) и ENSAT.
Всем пациентам с АКР показано регулярное проведение лабораторных тестов для определения гормональной опухолевой секреции, что способствует раннему выявлению рецидива/ прогрессирования заболевания. Очевидно, что биохимической оценке, в первую очередь, подлежат стероидные гормоны и/или метаболиты, которые присутствовали во время диагностики исходной опухоли. Тем не менее, ряд экспертов целесообразно оценивать весь стероидный спектр коры надпочечников, так как некоторые АКР могут с течением времени изменять функциональную активность [9].
5.2.2. Всем пациентам с АКР, получающим терапию митотаном**, рекомендуется регулярное определение концентрации митотана** в крови. Целевой диапазон концентрации митотана** в крови составляет 14 - 20 мкг/мл [8, 83, 102, 103] (см. также Приложение АЗ).
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 4
Комментарий. Узкий терапевтический диапазон митотана** обусловливает необходимость регулярного мониторирования его концентрации в плазме крови. Определение уровня митотана** в плазме оптимально проводить в утренние часы через, по крайней мере, 12 часов от момента последнего его приема.
На апарте или при возобновлении терапии митотаном** до тех пор, пока его концентрация не достигла целевого уровня > 14 мкг/мл, следует проводить мониторирование не реже 1 раза в 3-4 недели; в дальнейшем после достижения и стабильного сохранения терапевтической концентрации в крови достаточно измерять уровень митотан** в плазме каждые 4-12 недель.
На апарте или при возобновлении терапии митотаном** до тех пор, пока его концентрация <14 мкг/мл, целесообразно проводить постепенную эскалацию суточной дозы при нормальной переноашости до достижения терапевтического диапазона. При подборе оптимальной дозы митотана** необходимо учитывать особенности его
фармакокинетики, в частности, высокую липофильность, обусловливающую депонирование в жировой ткани и медленное высвобождение в кровь в дальнейшем.
Большинство пациентов отмечает те или иные побочные эффекты митотана** выраженность которых коррелирует, как правило, с его концентрацией в крови (см. также разд. 3.2.2 и табл. 6). Тем не менее, некоторые желудочно-кишечные побочные эффекты (такие как диарея), по-видимому, в большей степени коррелируют с пероральной дозировкой, чем с уровнем в плазме, и встречаются чаще в начале лечения [8, 83, 83, 102, 103]. Нежелательные явления неврологического шрактера чаще возникают при превышении концентрации 20 мкг/мл [76-78]. Поэтому многие эксперты рекомендуют поддерживать концентрацию в плазме ниже 20 мкг/мл. Однако есть данные о том, что более высокие уровни митотана** в плазме также могут быть связаны с более высокой эффективностью. Кроме того, некоторые пациенты не испытывают соответствующих нежелательных явлений даже при уровнях в плазме значительно выше 20 мкг/мл.
• 5.2.3. Всем пациентам с АКР, получающим терапию митотаном**, рекомендуется регулярное обследование на предмет нежелательных явлений, и своевременное их лечение [76-78].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 4
Комментарий. Помимо надпочечниковой недостаточности лечение митотаном** может сопровождаться множеством потенциальных нежелательных явлений (см также разд. 3.2.2 и табл. 6). (Табл. 2). Всем пациентам с АКР, получающим терапию митотаном** необходимо проводить регулярное обследование на предмет нежелательных явлений (Табл. 8). Важность своевременного их выявления и купирования очевидна [9].

Желудочно-кишечные побочные эффекты нередки, особенно в первые месяцы терапии
митотаном** Поддерживающая терапия должна включать нротиворвотные и противодиарейные препараты, если необходимо. Следует помнить, что тошнота/рвота также могут быть признаками надпочечниковой недостаточности, которая требует незамедлительного лечения. Важно отметить, что ключевым фактором, влияющим на достижения терапевтического диапазона митотана** в плазме, является именно переносимость терапии.
В случае развития умеренных побочных эффектов со стороны центральной нервной системы или ЖКТ дозу митотана** следует снизить на 0.5-1 г/сут и провести соответствующее симптоматическое лечение. В случае серьезных, но не жизнеугорожающих побочных эффектов со стороны ЦНС или любых жизнеугрожающих побочных эффектов и / или повышения уровня ферментов печени > в 5 раз, кроме ГГТП, необходимо временно прекратить прием митотана** и провести соответствующее симптоматическое лечение. Возобновление приема митотана** в редуцированной дозе (50-75% от последней дозы) возможно после купирования побочных эффектов под строгим контролем состояния пациента.
На фоне приема митотана** возможно развитие клинической картины центрального гипотиреоза, в связи с чем необходимо регулярное определение ТТГ, Т4 св. и проведение заместительной терапии левотироксином натрия** по показаниям.
Оценка уровней тестостерона и ГСПГ в динамике, а также при развитии клинической картины гипогонадизма, является оправданной у мужчин, получающих терапию митотаном**. В случае симптоматики гипогонадизма в сочетании со снижением тестостерона необходимо рассмотреть вопрос заместительной терапии.
Яичниковый стероидогенез, как и женская репродуктивная функция, в целом, менее подвержены влиянию митотана**. Однако у женщин репродуктивного возраста, получающих митотан**, могут наблюдаться множественные кисты яичников.
Гиперхолестеринемия и/или дислипидемия являются частыми нежелательными явлениями терапии митотаном**. Гиполипидемическая терапия с ипользованием статином, НЕ метаболизируемых CYP3A4), рекомендована пациентам, получающим митотан**, с учетом прогноза, кардиоваскулярных рисков и т.п.
Рекомендации по заместительной терапии см. также в Приложении АЗ.
5.5. Прогноз
• 5.3.1. Для оценки прогноза и определения оптимального лечебного алгоритма на момент постановки диагноза рекомендуется учитывать следующие факторы: стадия заболевания, резекционный статус, индекс Ki67, автономная секреция кортизола и общее состояние пациента [107-112,123,124].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 4
Комментарий. Прогноз АКР, в основном, зависит от стадии заболевания по ENSAT, так как она отражает распространенность опухолевого процесса: 5-летняя выживаемость при I стадии составляет 70-80%, при II стадии - 60-70%, при III - 20-40%, при IV - 0-10% соответственно. Общая, неранжированная по стадиям, пятилетняя выживаемость колеблется от 16 до 38%. Медиана общей выживаемости при IV стадии - менее 12 месяцев. [104-106].
По результатам ряда исследований [107-112], резекционный статус и пролиферативный индекс Ki67 были отнесены к важным прогностическим факторам. При распространенных формах заболевания (более 40% пациентов на момент первичной диагностики) значительное отрицательное влияние на сроки выживания оказывает синдром гиперкортицизма. Наконец, общее состояние пациента является очевидным прогностическим фактором, особенно в пожилом возрасте [112].
В 2015г. группой ENSAT проведен ретроспективный анализ прогностической значимости индекса Ki67 при АКР [123]. Отдельно анализировали немецкую группу от остальной как более гомогенную. Критические значения индекса Ki67 были приняты от 5 до 10%, 10-20% и более 20%. Как общая, так и безрецидивная выживаемость достоверно отличались как в германской, так и в общей группе при проведении однофакторного и многофакторного анализа. В этом исследовании другими достоверными прогностическими факторами оказались размер опухоли и инвазия венозных структур. На основании чего были сформированы группы риска: с индексом Ki67 больше 10% и больше 20%, наличие венозной инвазии или размер опухоли 15-20см приравнялись к третьей группе риска.
Международное исследование индекса Ki67 в 101 случае АКР с помощью традиционного метода подсчета, в том числе - пересмотр несколькими специалистами, а также с применением метода цифрового анализа изображений, показало, что имеется разброс результатов, и они зависят от метода. Авторы пришли к выводу, что при подсчете индекса Ki67 с применением цифрового анализа изображений, группы пациентов со значением индекса как 0-10-20%, так и 0-15-30% достоверно отличались по прогнозу, когда анализировали общую выживаемость [124].
При распространенном АКР (IV стадия по ENSAT, определяемая по наличию отдаленных метастазов, и/или рецидив заболевания, не подлежащий хирургическому лечению в объеме R0, и/ или Р2-резекционный статус) основными прогностическими факторами, связанными с худшим прогнозом, являются распространенность опухоли, высокий пролиферативный индекс Ki67 и плохо контролируемая опухолевая симптоматика.
• 5.3.2. В ходе динамического наблюдения рекомендуется пересматривать прогноз при каждом контрольном визите для определения оптимальной тактики лечения. [1, 8,9]
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 5
После проведения радикального хирургического лечения главным прогностическим фактором является факт рецидива опухоли. В случае его возникновения прогностическую роль играют время до прогрессирования, распространенность опухолевого процесса и резектабельность.
При метастатическом АКР, спектр прогностических факторов включает пролиферативный индекс Ki67, распространенность опухолевого процесса, общее состояние пациента, динамику прогрессирования, ответ на лечение.
Важно отметить, что оценка этих факторов в совокупности позволяет сделать лишь предварительные выводы; прогностические многофакторные системы до настоящего времени не разработаны.
5.4. АКР и беременность
• 5.4.1. При выявлении опухоли с высоким злокачественным потенциалом во время беременности, рекомендуется проведение хирургического лечения в кратчайшие сроки, вне зависимости от триместра беременности [114,115].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 4
Комментарий. Учитывая важность быстрого и радикального хирургического лечения для дальнейшего прогноза при АКР, оперативное лечение необходимо провести в кратчайшие сроки от постановки диагноза и независимо от срока беременности. При этом преждевременные роды (особенно в третьем триместре) и потеря беременности являются очевидными хирургическими рисками. Очевидно, что решение об оптимальной тактике лечения беременной пациентки с высоковероятным диагнозом АКР должно быть коллегиальным, с участием мулыпидисциплинарной команды. Оно должно учитывать прогноз заболевания и риск для матери и плода в связи с основным заболеванием и интервенционными процедурами. Пациентка должна быть информирована о возможных рисках и преимуществах хирургического лечения во время беременности и должна принять участие в информированном обсуждении.
На сегодняшний день нет убедительных данных относительно безопасности и оптимальных сроков наступления беременности при АКР. Важно отметить, что основной проблемой является в целом неблагоприятный прогноз при АКР, а также вероятность того, что беременность может увеличить риск рецидива. Имеются ограниченные данные о том, что АКР, выявленный во время беременности или в послеродовом периоде, имеет худший прогноз, чем у небеременных женщин [ИЗ]. Была выдвинута гипотеза о том, что беременность может способствовать развитию более агрессивного варианта АКР. В то же время Pauline de Corbiere и соавт. [114-119] не выявили негативного влияния беременности на клиническое течение и прогноз АКР у пациенток, предварительно получивших надлежащее лечение АКР. Ограничениями исследования являются малый объем выборки (17 пациенток, 21 беременность) и ретроспективный дизайн, не позволяющий обеспечить достаточную достоверность.
В связи с крайним дефицитом доказательной базы по проблеме беременности и АКР необходимо информировать пациенток о значительном риске рецидива АКР болезни в первые годы после постановки диагноза, а также о возможном прогрессировании АКР на фоне беременности. •
• 5.4.2. Наступление беременности на фоне терапии митотаном** противопоказано [9].
Уровень убедительности рекомендаций - С, уровень достоверности доказательств - 5
Комментарий. Проблема применения митотана** во время беременности связана с потенциальными тератогенными эффектами, которые могут быть обусловлены его проникновением через плаценту и адренолитическим эффектом на плод. На сегодняшний день опубликованы единичные сообщения о случаях беременности при приеме митотана**. Фактическое отсутствие доказательной базы не позволяет сделать окончательные выводы о безопасности лечения митотаном** во время беременности или связанных с ним рисках. Пациентка, получающая митотан** должна быть проинформирована об этих рисках и обеспечить эффективную контрацепцию.
Рассмотрение вопроса о планировании беременности возможно только после прекращения терапии митотаном** и снижения его концентрации в крови до неопределяемых значений (это может занять 3-12 месяцев).
В случае наступления беременности во время терапии митотаном** пациентка должна быть информирована о возможных рисках для плода, связанных с приемом митотана**, и возможных рисках прогрессирования АКР на фоне отмены митотана**. В случае, если пациентка желает сохранить беременность, лечение митотаном ** следует прекратить [9].
Организация оказания медицинской помощи
Медицинская помощь, за исключением медицинской помощи в рамках клинической апробации, в соответствии с федеральным законом от 21.11.2011 № 323-ФЗ (ред. От 25.05.2019) «Об основах охраны здоровья граждан в Российской Федерации», организуется и оказывается:
1) в соответствии с положением об организации оказания медицинской помощи по видам медицинской помощи, которое утверждается уполномоченным федеральным органом исполнительной власти;
2) в соответствии с порядком оказания помощи по профилю «онкология», обязательным для исполнения на территории Российской Федерации всеми медицинскими организациями;
3) на основе настоящих клинических рекомендаций;
4) с учетом стандартов медицинской помощи, утвержденных уполномоченным федеральным органом исполнительной власти.
Первичная специализированная медико-санитарная помощь оказывается врачом-онкологом и иными врачами-специалистами в центре амбулаторной онкологической помощи либо в первичном онкологическом кабинете, первичном онкологическом отделении, поликлиническом отделении онкологического диспансера.
При подозрении или выявлении у пациента онкологического заболевания врачи-терапевты, врачи-терапевты участковые, врачи общей практики (семейные врачи), врачи-специалисты, средние медицинские работники в установленном порядке направляют пациента на консультацию в центр амбулаторной онкологической помощи либо в первичный онкологический кабинет, первичное онкологическое отделение медицинской организации для оказания ему первичной специализированной медико-санитарной помощи.
Консультация в центре амбулаторной онкологической помощи либо в первичном онкологическом кабинете, первичном онкологическом отделении медицинской организации должна быть проведена не позднее 5 рабочих дней с даты выдачи направления на консультацию. Врач-онколог центра амбулаторной онкологической помощи (в случае отсутствия центра амбулаторной онкологической помощи врач-онколог первичного онкологического кабинета или первичного онкологического отделения) организует взятие биопсийного (операционного) материала, а также организует выполнение иных диагностических исследований, необходимых для установления диагноза, включая распространенность онкологического процесса и стадию заболевания.
В случае невозможности взятия в медицинской организации, в составе которой организован центр амбулаторной онкологической помощи (первичный онкологический кабинет, первичное онкологическое отделение), биопсийного (операционного) материала, проведения иных диагностических исследований пациент направляется лечащим врачом в онкологический диспансер или в медицинскую организацию, оказывающую медицинскую помощь пациентам с онкологическими заболеваниями.
Срок выполнения патолого-анатомических исследований, необходимых для гистологической верификации злокачественного новообразования, не должен превышать 15 рабочих дней с даты поступления биопсийного (операционного) материала в патолого-анатомическое бюро (отделение).
При подозрении и (или) выявлении у пациента онкологического заболевания в ходе оказания ему скорой медицинской помощи таких пациентов переводят или направляют в медицинские организации, оказывающие медицинскую помощь пациентам с онкологическими заболеваниями, для определения тактики ведения и необходимости применения дополнительно других методов специализированного противоопухолевого лечения.
Врач-онколог центра амбулаторной онкологической помощи (первичного онкологического кабинета, первичного онкологического отделения) направляет пациента в онкологический диспансер или в медицинские организации, оказывающие медицинскую помощь пациентам с онкологическими заболеваниями, для уточнения диагноза (в случае невозможности установления диагноза, включая распространенность онкологического процесса и стадию заболевания, врачом-онкологом центра амбулаторной онкологической помощи, первичного онкологического кабинета или первичного онкологического отделения) и оказания специализированной, в том числе высокотехнологичной, медицинской помощи.
Срок начала оказания специализированной, за исключением высокотехнологичной, медицинской помощи пациентам с онкологическими заболеваниями в медицинской организации, оказывающей медицинскую помощь пациентам с онкологическими заболеваниями, не должен превышать 14 календарных дней с даты гистологической верификации злокачественного новообразования или 14 календарных дней с даты установления предварительного диагноза злокачественного новообразования (в случае отсутствия медицинских показаний для проведения патолого-анатомических исследований в амбулаторных условиях).
Специализированная, в том числе высокотехнологичная, медицинская помощь оказывается врачами-онкологами, врачами-радиотерапевтами в онкологическом диспансере или в медицинских организациях, оказывающих медицинскую помощь пациентам с онкологическими заболеваниями, имеющих лицензию, необходимую материально-техническую базу, сертифицированных специалистов, в стационарных условиях и условиях дневного стационара и включает в себя профилактику, диагностику, лечение онкологических заболеваний, требующих использования специальных методов и сложных уникальных медицинских технологий, а также медицинскую реабилитацию.
В медицинской организации, оказывающей медицинскую помощь пациентам с онкологическими заболеваниями, тактика медицинского обследования и лечения устанавливается консилиумом врачей-онкологов и врачей-радиотерапевтов, с привлечением при необходимости других врачей-специалистов. Решение консилиума врачей оформляется протоколом, подписывается участниками консилиума врачей и вносится в медицинскую документацию пациента.
Показания для госпитализации в круглосуточный или дневной стационар медицинской организации, оказывающей специализированную, в том числе высокотехнологичную медицинскую помощь по профилю «онкология» определяются консилиумом врачей-онкологов и врачей-радиотерапевтов, с привлечением при необходимости других врачей-специалистов.
Показанием для госпитализации в медицинскую организацию в экстренной или неотложной форме является:
1) наличии осложнений онкологического заболевания, требующих оказания ему специализированной медицинской помощи в экстренной и неотложной форме;
2) наличие осложнений лечения (хирургическое вмешательство, лучевая терапия, лекарственная терапия и т.д.) онкологического заболевания.
Показанием для госпитализации в медицинскую организацию в плановой форме является:
1) необходимость выполнения сложных интервенционных диагностических медицинских вмешательств, требующих последующего наблюдения в условиях круглосуточного или дневного стационара;
2) наличие показаний к специализированному противоопухолевому лечению (хирургическое вмешательство, лучевая терапия, в том числе контактная, дистанционная и другие виды лучевой терапии, лекарственная терапия и др.), требующему наблюдения в условиях круглосуточного или дневного стационара.
Показанием к выписке пациента из медицинской организации является:
1) завершение курса лечения, или одного из этапов оказания специализированной, в том числе высокотехнологичной медицинской помощи, в условиях
круглосуточного или дневного стационара при условиях отсутствия осложнений лечения, требующих медикаментозной коррекции и/или медицинских вмешательств в стационарных условиях;
2) отказ пациента или его законного представителя от специализированной, в том числе высокотехнологичной медицинской помощи в условиях круглосуточного или дневного стационара, установленной консилиумом медицинской организации, оказывающей онкологическую помощь при условии отсутствия осложнений основного заболевания и/или лечения, требующих медикаментозной коррекции и/или медицинских вмешательств в стационарных условиях;
3) необходимость перевода пациента в другую медицинскую организацию по соответствующему профилю оказания медицинской помощи.
Заключение о целесообразности перевода пациента в профильную медицинскую организацию осуществляется после предварительной консультации по предоставленным медицинским документам и/или предварительного осмотра пациента врачами специалистами медицинской организации, в которую планируется перевод.
6. Дополнительная информация, влияющая на течение и исход заболевания
Факторы, влияющие на течение и исход АКР, включают следующие:
1. Распространенность опухолевого процесса;
2. Локализация метастатических очагов;
3. Биологические особенности опухоли;
4. Развитие нежелательных явлений при приеме препаратов для противоопухолевой терапии.
Критерии оценки качества медицинской помощи

Список литературы
1. Мельниченко Г.А., Стилиди И.С., Алексеев Б.Я., Горбунова В.А., Бельцевич Д.Г., Райхман А.О., Кузнецов Н.С., Жуков Н.В., Бохян В.Ю. Федеральные клинические рекомендации по диагностике и лечению адренокортикального рака. Проблемы эндокринологии. 2014; 60(2): 51-67. doi: 10.14341/ргоЫ201460251-67
2. Lloyd RV, Osamura RY, Kloppel G, Rosai J, eds. WHO Classification of Tumours of Endocrine Organs. Fourth Edition. Lyon: IARC; 2017
3. Селиванова Л.С., Рослякова A.A., Боголюбова A.B., Тертычный А.С., Бельцевич Д.Г., Абросимов А.Ю., Мельниченко Г.А. Молекулярно-генетические маркеры и критерии прогноза адренокортикального рака. Архив патологии. 2019;81(5):92-96. doi: 10.17116/patol20198105192
4. F.lseT. Association of Adrenocortical Carcinoma with Familial Cancer Susceptibility Syndromes. Mol Cell Endocrinol. 2012: 351П1: 66-70. doi: 10.1016/i.mce.2Q11.12.008
2. Lloyd RV, Osamura RY, Kloppel G, Rosai J, eds. WHO Classification of Tumours of Endocrine Organs. Fourth Edition. Lyon: IARC; 2017
3. Селиванова Л.С., Рослякова A.A., Боголюбова A.B., Тертычный А.С., Бельцевич Д.Г., Абросимов А.Ю., Мельниченко Г.А. Молекулярно-генетические маркеры и критерии прогноза адренокортикального рака. Архив патологии. 2019;81(5):92-96. doi: 10.17116/patol20198105192
4. F.lseT. Association of Adrenocortical Carcinoma with Familial Cancer Susceptibility Syndromes. Mol Cell Endocrinol. 2012: 351П1: 66-70. doi: 10.1016/i.mce.2Q11.12.008
5. Schteineart DE. Doherty CM. Gauger PC. et al. Management of patients with adrenal cancer: recommendations of an international consensus conference. Endocr Relat Cancer. 2005:12:667. doi: 10.1677/erc. 1.01029
6. Министерство здравоохранения Российской Федерации. Перечень редких (орфанных! заболеваний
7. Adrenal Cortical Carcinoma. In: Brierlev ID. Gospodarowicz MK. Wittekind C feds'). TNM Classification of Malignant Tumours 18th edition). Oxford. UK: Wilev-Blackwell. 2017
8. Fassnacht M. Dekkers O. Else T. Baudin E. Berruti A. de Kruger RR. Haak HR. Mihai R. Assie G. Terzolo M. European Society of Endocrinology Clinical Practice Guidelines on the Management of Adrenocortical Carcinoma in Adults, in collaboration with the European Network for the Study of Adrenal Tumors. F.ur 1 Endocrinol. F.1E. 2018: 179641: C1-C46. doi: 10.1550/F.1F.-18-0608.
9. Fassnacht M. Allolio B. Clinical management of adrenocortical carcinoma. Best Pract Res ClinEndocrinolMetab. 2009:23121:273-89. doi: 10.1016/i.beem.2008.10.008
10. Chuang B. Fassnacht M. Adrenocortical carcinoma: clinical update. 1 ClinEndocrinolMetab. 2006:91f6'):2027-37. doi: 10.1210/ic.2005-2639
11. Terzolo M. Ali A. Osella G. Mazza E. Prevalence of adrenal carcinoma among incidentally discovered adrenal masses. A retrospective study from 1989 to 1994. Gruppo Piemontese Incidentalomi Surrenalici. Arch Surg. 1997:
12. Cawood TL Hunt PL O’Shea D. Cole D. Soule S. Recommended evaluation of adrenal incidentalomas is costly, has high false-positive rates and confers a risk of fatal cancer that is similar to the risk of the adrenal lesion becoming malignant: time for a rethink? Eur 1 Endocrinol. 2009:161(49:513-27. doi: 10.1530/E1E-09-0234.
13. Fassnacht M. Arlt W. Bancos I. Dralle H. Newell-Price 1. Sahdev A. Tabarin A. Terzolo M. Tsagarakis S. Dekkers OM. Management of adrenal incidentalomas: European Society of Endocrinology Clinical Practice Guideline in collaboration with the European Network for the Study of Adrenal Tumors. Eur 1 Endocrinol. 2016:175f2f:Gl-G34. doi: 10.1530/E1E-16-0467.
14. Селиванова Л.С.. Рослякова A.A.. Коваленко Ю.А.. Боголюбова А.В.. Тертычный А.С.. Бельцевич Д.Г.. Абросимов А.Ю.. Мельниченко Г.А. Современные критерии диагностики алренокортикального рака. Архив патологии. 2019:81131:66-73.
15. Белытевич Л.Г.. Кузнецов Н.С.. Соллатова Т.В.. Ванутпко В.Э. Инттиленталома надпочечников. Эндокринная хирургия. 2009:3111:19-23.
16. Bovio S. Cataldi A. Reimondo G. et al. Prevalence of adrenal incidentaloma in a contemporary computerized tomography series. 1 Endocrinol Invest. 2006:29141:298-302. doi: 10.1007/BF03344099
17. Young WFlr. The Incidentally Discovered Adrenal Mass. N Engl 1 Med. 2007:356:601-10. doi: IO. 1056/NElMcp065470
18. Libe R. DallAsta C. Barbetta L et al. Long-term follow-up study of patients with adrenal incidentalomas. Eur I Endocrinol. 2002:147:489-494. doi: 10.1550/eie.Q. 1470489
19. Terzolo M. Reimondo C. Bovio S. Angeli A Suhclinical Cushing’s syndrome. Pituitary. 2004:7С41:217-223. doi: 10.1007/s11102-005-4024-6
20. Kirkhv-Bott I. Brunaud L. Mathonet M. Harnoir E. Krainips TL. Tresallet C. Arnar L. Rault A. Henry JF. Carnaille B. Ectopic hormone-secreting pheochromocvtoma: a francophone observational study. World T Surg. 2012:36161:1382-8. doi: 10.1007/s00268-012-1488-l.
21. Li XG. Zhang DX. Li X. Cui XG. Xu DF. Li Y. Gao Y. Yin L. Ren TZ Adrenocorticotropic hormone-producing pheochromocvtoma: a case report and review of the literature. Chin Med I ('Engl). 2012:12516'): 1193-1196. doi: 10.5760/cma.i.issn.0366-6999.2012.06.042
22. Cohade C. Broussaud S. Louiset E. Bennet A. Huvghe E. Caron P. Ectopic Cusliing"s syndrome due to a pheochromocvtoma: a new case in the post-partum and review of literature. Gvnecol Endocrinol. 2009:25191:624-627. doi: 10.1080/09513590903015411
23. Gardet V et al. Lessons from an unpleasant surprise: a biochemical strategy for the diagnosis of pheochromocvtoma. I Hvpertens. 2001:19161:1029-1035.10.1097/00004872-200106000-00006
24. Niernan LK. Biller BMK. Findling TW et al. The diagnosis of Cushing’s syndrome: an endocrine society clinical practice guideline. I Clin Endocrinol Metab. 2008:93151:1526-40. doi: 10.1210/ic. 2008-0125.
25. Newell-Price I. Trainer P. Besser M. Grossman A. The diagnosis and differential diagnosis of Cushing’s svndronre and pseudo-Cushing"s states. Endocr Rev. 1998:19151:647-672. doi: 10.1210/edrv. 19.5.0346
26. Pecori Giraldi F. Ambrogio AG. De Martin M. et al. Specificity of first-line tests for the diagnosis of Cushing’s svndronre: assessment in a large series. I Clin Endocrinol Metab. 2007:921111:4123-9. doi: 10.1210/ic.2007-0596
27. Tsagarakis S. Vassiliadi D. Thalassinos N. Endogenous suhclinical hvpercortisolism: diagnostic uncertainties and clinical implications. I Endocrinol Invest. 2006:2915'):471 -82. doi: 10.1007/BF03344133
28. Mitchell IC. Auchus RT. Tuneia K. et al. “Suhclinical Cushing’s svndronre” is not suhclinical: improvement after adrenalectomy in 9 patients. Surgery 2007:142161:900-905. doi: 10.1016/i.surg.2007.10.001
29. Barzon L. Fallo F. Sonino N. Boscaro M. Development of overt Cushing’s svndronre in patients with adrenal incidentalonra. Eur 1 Endocrinol. 2002:14611'):61-66. doi: 0.1530/eie.0.1460061
30. Reincke M. Suhclinical Cushing’s svndronre. Endocrinol Metab Clin North Anr. 2000:29111:43-56. doi: 10.1016/80889-8529105170115-8
31. Pacak K. Preoperative management of the pheochromocvtoma patient. 1 Clin Endocrinol Metab 2007:92:4069-4079. doi: 10.1210/ic.2007-1720
32. Williams DT. Dann S. Wheeler MH. Phaeochromocvtoma - views on current management. Eur 1 Surg Oncol. 2003:29161:483-490
33. Kinnev M. Narr BL Warner MA. Perioperative management of pheochromocvtoma. 1 Cardiothorac Vase Anesth. 2002:16:359-369. doi: 10.1053/ican.2002.124150
34. Pacak К et al. Recent advances in genetics, diagnosis, localization, and treatment of pheochromocvtoma. Ann Intern Med. 2001:134141:315-329. doi: 10.7326/0003-4819-134-4-200102200-00016
35. Дедов И.И.. Бельпевич Д.Е.. Кузнецов H.C.. Мельниченко Е.А. Феохромопитома. Москва: Практ. медицина: 2005:47-70.
36. An Endocrine Society Clinical Practice Guidelines. Case Detection. Diagnosis, and Treatment of Patients with Primary Aldosteronism. 1 Clin Endocrinol Metab. 2008:93191:3266-3281.
37. Мельниченко Г.А.. Платонова H.M.. Бельпевич П.Г. и др. Первичный гиперальдостеронизм: диагностика и лечение. Новый взгляд на проблему. По материалам Проекта клинических рекомендаций Российской ассоциации эндокринологов по диагностике и лечению первичного гиперальдостеронизма. Consilium Medicum. 2017:19141: 75-85.
38. Mantero F. Terzolo M. Arnaldi G. et al. A survey on adrenal incidentalonra in Italy. Study Group on Adrenal Tumors of the Italian Society of Endocrinology. 1 Clin Endocrinol Metab. 2000:85121:637-644.
39. Angeli A. Osella G. Ali A. Terzolo M. Adrenal incidentalonra: an overview of clinical and epidemiological data from the National Italian Study Group. Hornr Res. 1997:47:279-283.
40. Metser IJ. Miller E. Lernian H et al. 18F-FDG PF.T/CT in the Evaluation of Adrenal Masses. 1 Nucl Med. 2006:47111:32-37.
41. M. Blake. P. Prakash. C.Cronin. PET/CT for Adrenal Assessment Am 1 Roentgenology. 2010:195121:195.
42. Mackie GC. Shulkin BL. Ribeiro RC. et al. Use of [18F]fluorodeoxvglucose positron emission tomography in evaluating locally recurrent and metastatic adrenocortical carcinoma. T Clin Endocrinol Metab. 2006:91:2665.
43. Leboulleux S. Dronrain C. Bonniaud G et al. Diagnostic and prognostic value of 18-fluorodeoxvglucose positron emission tomography in adrenocortical carcinoma: a prospective comparison with computed tomography. T Clin Endocrinol Metab. 2006:91:920.
44. Deandreis D. Leboulleux S. Caramella C. et al. FDG PET in the management of patients with adrenal masses and adrenocortical carcinoma. Horm Cancer. 2011:2:354.
45. Welch Tl. Sheedv PF II. Stephens DH et al. Percutaneous adrenal biopsv: review of a 10-vear experience. Radiology. 1994:195:541-544
46. Pacak K. Eisenhofer G. Ahlnran H et al. Pheochronrocvtonra: recommendations for clinical practice from the First International Symposium. Nat Clin Pract Endocrinol Metab. 2007:5:92-102
47. Dackiw АР. I,ее IF,. Gagel RF. F.vans DB. Adrenal cortical carcinoma. World 1 Surg. 2001:25:914-926
48. Ne h. T.ihertino 1M. Adrenocortical carcinoma: diagnosis, evaluation and treatment. 1 IJrol. 2005:169:5-11
49. Weiss LM. Comparative histologic study of 45 metastasizing and nonmetastasizing adrenocortical tumors. Am 1 Surg Pathol. 1984:8t51:165-169.
50. Lau SK. Weiss LM. The Weiss system for evaluating adrenocortical neoplasms: 25 years later. Hum Pathol. 2009:40161:75 7-768.
51. Van Slooten H. Schaberg A. Snreenk D. Moolenar Al. Morphologic characteristics of benign and malignant adrenocortical tumors. Cancer. 1985 Feb 15:55141:766-775.
52. Blanes A et al. Histologic criteria for adrenocortical proliferative lesions: value of mitotic figure variability. Am 1 Clin Pathol. 2007:127151:598-408.
53. Филиппова OB. А аренокортикальный рак: клинические проявления и морфологическая лиагностика / Филиппова О.В.. Хмельнитткая Н.М. // Мелиттинский альманах. 2011:5:115-
54. Wangberg В. Khorranr-Manesh A. lansson S. et al. The long-term survival in adrenocortical carcinoma with active surgical management and use of monitored nritotane. Endocr Relat Cancer. 2010:17:265.
55. Филимонюк A.B.. Харченко H.B.. Леонов Б.И.. Смирнова Е.А.. Антонов А.К.. Смелкова Н.И.. Непосредственные и отдаленные результаты хирургического лечения больных адренокортикальным раком. Вестник новых медицинских технологий (электронное издание). 2015:1 [Электронный ресурс].
56. Casola G. Nicolet У. van Sonnenherg F, et al. Unsuspected pheochromocvtoma: risk of blood-pressure alterations during percutaneous adrenal biopsy. Radiology. 1986:159:755-755.
57. Reihetanz 1. lurowich C. F.rdogan 1 et al. Impact of lvmphadenectomv on the oncologic outcome of patients with adrenocortical carcinoma. Ann Surg. 2012:255:565
58. Gauioux S. Brennan MF. Recommendation for standardized surgical management of primary adrenocortical carcinoma. Surgery. 2012 152111:125-52
59. Leboulleux S. Deandreis D. Ghuzlan A Al et al. Adrenocortical carcinoma: is the surgical approach a risk factor of peritoneal carcinomatosis? Eur 1 Endocrinol. 2010:162:1147-1155
60. Miller BS. Gauger PG. Hammer GD et al. Proposal for modification of the ENSAT staging system for adrenocortical carcinoma using tumor grade. Langenbecks Arch Surg. 2010:595:955/
61. Shen WT. Sturgeon C. Duh OY. From incidentaloma to adrenocortical carcinoma: the surgical management of adrenal tumors. 1 Surg Oncol. 2005:89151:186-92. doi: 10.1002/iso.2Q180
62. Schulick RD. Brennan MF. Long-term survival after complete resection and repeat resection in patients with adrenocortical carcinoma. Ann Surg Oncol. 1999:6181:719-726
65. Icard P. Chapuis Y. Andreassian B. et al. Adrenocortical carcinoma in surgically treated patients: a retrospective study on 156 cases bv the French Association of Endocrine Surgery. Surgery. 1992:112161:972-979.
64. Bellantone R. Ferrante A. Boscherini M. et al.: Role of reoperation in recurrence of adrenal cortical carcinoma: results from 188 cases collected in the Italian National Registry for Adrenal Cortical Carcinoma. Surgery. 1997.122:1212-1218.
65. Porpiglia F. Fiori C. Scarpa RM et al. The role of surgery in the management of recurrent adrenocortical carcinoma: results of a retrospective study. F.ur Urol Suppl. 2009:8(4):505.
66. Brix D. Allolio B. Fenske W et al. Laparoscopic versus open adrenalectomy for adrenocortical carcinoma: surgical and oncologic outcome in 152 patients. F.ur IJrol. 2010:58:609.
67. Hahner S. Fassnacht M. Mitotane for adrenocortical carcinoma treatment. Curr Opin Investig Drugs. 2005:6:586-594.
68. Schteingart DE. Conventional and novel strategies in the treatment of adrenocortical cancer. Braz 1 Med Biol Res. 2000:55:1197-1200
69. Hague RV. Mav W. Cullen DR. Hepatic microsomal enzvnre induction and adrenal crisis due to o.p DDD therapy for metastatic adrenocortical carcinoma. Clin Endocrinol ЮхГ). 1989:51:51-57
70. Министерство здравоохранения Российской Федерации. Перечень жизненно необходимых и важнейших лекарственных препаратов для медицинского применения.
study. World T Surg. 2012:36161:1382-8. doi: 10.1007/s00268-012-1488-l.
71. Li XG. Zhang DX. Li X. Cui XG. Xu DF. Li Y. Gao Y. Yin L. Ren TZ Adrenocorticotropic hormone-producing pheochromocvtoma: a case report and review of the literature. Chin Med I ('Engl). 2012:12516'): 1193-1196. doi: 10.5760/cma.i.issn.0366-6999.2012.06.042
72. Cohade C. Broussaud S. Louiset E. Bennet A. Huvghe E. Caron P. Ectopic Cusliing"s syndrome due to a pheochromocvtoma: a new case in the post-partum and review of literature. Gvnecol Endocrinol. 2009:25191:624-627. doi: 10.1080/09513590903015411
73. Gardet V et al. Lessons from an unpleasant surprise: a biochemical strategy for the diagnosis of pheochromocvtoma. I Hvpertens. 2001:19161:1029-1035.10.1097/00004872-200106000-00006
74. Niernan LK. Biller BMK. Findling TW et al. The diagnosis of Cushing’s syndrome: an endocrine society clinical practice guideline. I Clin Endocrinol Metab. 2008:93151:1526-40. doi: 10.1210/ic. 2008-0125.
25. Newell-Price I. Trainer P. Besser M. Grossman A. The diagnosis and differential diagnosis of Cushing’s svndronre and pseudo-Cushing"s states. Endocr Rev. 1998:19151:647-672. doi: 10.1210/edrv. 19.5.0346
26. Pecori Giraldi F. Ambrogio AG. De Martin M. et al. Specificity of first-line tests for the diagnosis of Cushing’s svndronre: assessment in a large series. I Clin Endocrinol Metab. 2007:921111:4123-9. doi: 10.1210/ic.2007-0596
27. Tsagarakis S. Vassiliadi D. Thalassinos N. Endogenous suhclinical hvpercortisolism: diagnostic uncertainties and clinical implications. I Endocrinol Invest. 2006:2915'):471 -82. doi: 10.1007/BF03344133
28. Mitchell IC. Auchus RT. Tuneia K. et al. “Suhclinical Cushing’s svndronre” is not suhclinical: improvement after adrenalectomy in 9 patients. Surgery 2007:142161:900-905. doi: 10.1016/i.surg.2007.10.001
29. Barzon L. Fallo F. Sonino N. Boscaro M. Development of overt Cushing’s svndronre in patients with adrenal incidentalonra. Eur 1 Endocrinol. 2002:14611'):61-66. doi: 0.1530/eie.0.1460061
30. Reincke M. Suhclinical Cushing’s svndronre. Endocrinol Metab Clin North Anr. 2000:29111:43-56. doi: 10.1016/80889-8529105170115-8
31. Pacak K. Preoperative management of the pheochromocvtoma patient. 1 Clin Endocrinol Metab 2007:92:4069-4079. doi: 10.1210/ic.2007-1720
32. Williams DT. Dann S. Wheeler MH. Phaeochromocvtoma - views on current management. Eur 1 Surg Oncol. 2003:29161:483-490
33. Kinnev M. Narr BL Warner MA. Perioperative management of pheochromocvtoma. 1 Cardiothorac Vase Anesth. 2002:16:359-369. doi: 10.1053/ican.2002.124150
34. Pacak К et al. Recent advances in genetics, diagnosis, localization, and treatment of pheochromocvtoma. Ann Intern Med. 2001:134141:315-329. doi: 10.7326/0003-4819-134-4-200102200-00016
35. Дедов И.И.. Бельпевич Д.Е.. Кузнецов H.C.. Мельниченко Е.А. Феохромопитома. Москва: Практ. медицина: 2005:47-70.
36. An Endocrine Society Clinical Practice Guidelines. Case Detection. Diagnosis, and Treatment of Patients with Primary Aldosteronism. 1 Clin Endocrinol Metab. 2008:93191:3266-3281.
37. Мельниченко Г.А.. Платонова H.M.. Бельпевич П.Г. и др. Первичный гиперальдостеронизм: диагностика и лечение. Новый взгляд на проблему. По материалам Проекта клинических рекомендаций Российской ассоциации эндокринологов по диагностике и лечению первичного гиперальдостеронизма. Consilium Medicum. 2017:19141: 75-85.
38. Mantero F. Terzolo M. Arnaldi G. et al. A survey on adrenal incidentalonra in Italy. Study Group on Adrenal Tumors of the Italian Society of Endocrinology. 1 Clin Endocrinol Metab. 2000:85121:637-644.
39. Angeli A. Osella G. Ali A. Terzolo M. Adrenal incidentalonra: an overview of clinical and epidemiological data from the National Italian Study Group. Hornr Res. 1997:47:279-283.
40. Metser IJ. Miller E. Lernian H et al. 18F-FDG PF.T/CT in the Evaluation of Adrenal Masses. 1 Nucl Med. 2006:47111:32-37.
41. M. Blake. P. Prakash. C.Cronin. PET/CT for Adrenal Assessment Am 1 Roentgenology. 2010:195121:195.
42. Mackie GC. Shulkin BL. Ribeiro RC. et al. Use of [18F]fluorodeoxvglucose positron emission tomography in evaluating locally recurrent and metastatic adrenocortical carcinoma. T Clin Endocrinol Metab. 2006:91:2665.
43. Leboulleux S. Dronrain C. Bonniaud G et al. Diagnostic and prognostic value of 18-fluorodeoxvglucose positron emission tomography in adrenocortical carcinoma: a prospective comparison with computed tomography. T Clin Endocrinol Metab. 2006:91:920.
44. Deandreis D. Leboulleux S. Caramella C. et al. FDG PET in the management of patients with adrenal masses and adrenocortical carcinoma. Horm Cancer. 2011:2:354.
45. Welch Tl. Sheedv PF II. Stephens DH et al. Percutaneous adrenal biopsv: review of a 10-vear experience. Radiology. 1994:195:541-544
46. Pacak K. Eisenhofer G. Ahlnran H et al. Pheochronrocvtonra: recommendations for clinical practice from the First International Symposium. Nat Clin Pract Endocrinol Metab. 2007:5:92-102
47. Dackiw АР. I,ее IF,. Gagel RF. F.vans DB. Adrenal cortical carcinoma. World 1 Surg. 2001:25:914-926
48. Ne h. T.ihertino 1M. Adrenocortical carcinoma: diagnosis, evaluation and treatment. 1 IJrol. 2005:169:5-11
49. Weiss LM. Comparative histologic study of 45 metastasizing and nonmetastasizing adrenocortical tumors. Am 1 Surg Pathol. 1984:8t51:165-169.
50. Lau SK. Weiss LM. The Weiss system for evaluating adrenocortical neoplasms: 25 years later. Hum Pathol. 2009:40161:75 7-768.
51. Van Slooten H. Schaberg A. Snreenk D. Moolenar Al. Morphologic characteristics of benign and malignant adrenocortical tumors. Cancer. 1985 Feb 15:55141:766-775.
52. Blanes A et al. Histologic criteria for adrenocortical proliferative lesions: value of mitotic figure variability. Am 1 Clin Pathol. 2007:127151:598-408.
55. Филиппова OB. А аренокортикальный рак: клинические проявления и морфологическая лиагностика / Филиппова О.В.. Хмельнитткая Н.М. // Мелиттинский альманах. 2011:5:115-
116.
54. Wangberg В. Khorranr-Manesh A. lansson S. et al. The long-term survival in adrenocortical carcinoma with active surgical management and use of monitored nritotane. Endocr Relat Cancer. 2010:17:265.
55. Филимонюк A.B.. Харченко H.B.. Леонов Б.И.. Смирнова Е.А.. Антонов А.К.. Смелкова Н.И.. Непосредственные и отдаленные результаты хирургического лечения больных адренокортикальным раком. Вестник новых медицинских технологий (электронное издание). 2015:1 [Электронный ресурс].
56. Casola G. Nicolet У. van Sonnenherg F, et al. Unsuspected pheochromocvtoma: risk of blood-pressure alterations during percutaneous adrenal biopsy. Radiology. 1986:159:755-755.
57. Reihetanz 1. lurowich C. F.rdogan 1 et al. Impact of lvmphadenectomv on the oncologic outcome of patients with adrenocortical carcinoma. Ann Surg. 2012:255:565
58. Gauioux S. Brennan MF. Recommendation for standardized surgical management of primary adrenocortical carcinoma. Surgery. 2012 152111:125-52
59. Leboulleux S. Deandreis D. Ghuzlan A Al et al. Adrenocortical carcinoma: is the surgical approach a risk factor of peritoneal carcinomatosis? Eur 1 Endocrinol. 2010:162:1147-1155
60. Miller BS. Gauger PG. Hammer GD et al. Proposal for modification of the ENSAT staging system for adrenocortical carcinoma using tumor grade. Langenbecks Arch Surg. 2010:595:955/
61. Shen WT. Sturgeon C. Duh OY. From incidentaloma to adrenocortical carcinoma: the surgical management of adrenal tumors. 1 Surg Oncol. 2005:89151:186-92. doi: 10.1002/iso.2Q180
62. Schulick RD. Brennan MF. Long-term survival after complete resection and repeat resection in patients with adrenocortical carcinoma. Ann Surg Oncol. 1999:6181:719-726
65. Icard P. Chapuis Y. Andreassian B. et al. Adrenocortical carcinoma in surgically treated patients: a retrospective study on 156 cases bv the French Association of Endocrine Surgery. Surgery. 1992:112161:972-979.
64. Bellantone R. Ferrante A. Boscherini M. et al.: Role of reoperation in recurrence of adrenal cortical carcinoma: results from 188 cases collected in the Italian National Registry for Adrenal Cortical Carcinoma. Surgery. 1997.122:1212-1218.
65. Porpiglia F. Fiori C. Scarpa RM et al. The role of surgery in the management of recurrent adrenocortical carcinoma: results of a retrospective study. F.ur Urol Suppl. 2009:8(4):505.
66. Brix D. Allolio B. Fenske W et al. Laparoscopic versus open adrenalectomy for adrenocortical carcinoma: surgical and oncologic outcome in 152 patients. F.ur IJrol. 2010:58:609.
67. Hahner S. Fassnacht M. Mitotane for adrenocortical carcinoma treatment. Curr Opin Investig Drugs. 2005:6:586-594.
68. Schteingart DE. Conventional and novel strategies in the treatment of adrenocortical cancer. Braz 1 Med Biol Res. 2000:55:1197-1200
69. Hague RV. Mav W. Cullen DR. Hepatic microsomal enzvnre induction and adrenal crisis due to o.p DDD therapy for metastatic adrenocortical carcinoma. Clin Endocrinol ЮхГ). 1989:51:51-57
70. Министерство здравоохранения Российской Федерации. Перечень жизненно необходимых и важнейших лекарственных препаратов для медицинского применения.
71. Lim МС. Tan Y0. Chong PY. Cheah IS. Treatment of adrenal cortical carcinoma with mitotane: outcome and complications. Ann Acad Med Singapore. 1990.
72. Decker RA. Kuehner ME. Adrenocortical carcinoma. Am Surg. 1991:57:502-515.
73. Terzolo. M. Adjuvant mitotane treatment for adrenocortical carcinoma / M. Terzolo Angeli A. Fassnacht M et al. //N. Engl. I. Med. - 2007. - Vol. 556. №25. - P. 2572-2580.
74. Tlias I. Alevizaki M. Philippou G. Anastasiou F,. Souvatzoglou A. Sustained remission of metastatic adrenal carcinoma during long-term administration of low-dose mitotane. I Endocrinol Invest. 2001:24:532-535.
75. Allolio B. Hahner S. Weismann D. Fassnacht M. Management of adrenocortical carcinoma. Clin Endocrinol ЮхГ). 2004:60:273-287.19:540-544.
76. Hahner S. Fassnacht M. Mitotane for adrenocortical carcinoma treatment. Curr Opin Investig Drugs. 2005:6:386-394
77. Van Slooten H. Moolenaar AT. van Seters AP. Snreenk D. The treatment of adrenocortical carcinoma with o.p-DDD: prognostic implications of serum level monitoring. Eur I Cancer Clin Oncol. 1984:20:47-53.
78. Haak HR. Hermans I. van de Velde CT et al. Optimal treatment of adrenocortical carcinoma with mitotane: results in a consecutive series of 96 patients. Br I Cancer. 1994:69:947-951.
79. Baudin E. Pellegriti G. Bonnav M et al. Impact of monitoring plasma 1.1- dichlorodiphenildichloroethane (o.p DDD1 levels on the treatment of patients with adrenocortical carcinoma. Cancer. 2001:92:1385-1392.
80. Heilnrann P. Wagner P. Nawroth PP. Ziegler R [Therapy of the adrenocortical carcinoma with Lvsodren to.p’-DDD'). Therapeutic management bv monitoring o.p -DDD blood levels], Med Klin. 2001:96:371-377
81. Becker D. Schumacher OP. o.p’DDD therapy in invasive adrenocortical carcinoma. Ann Intern Med. 1975:82:677-679.
82. Boven E. Verniorken ТВ. van Slooten H. PinedoHM. Complete response of metastasized adrenal cortical carcinoma with o.p -DDD. Case report and literature review. Cancer. 1984:55:26-29.
83. Krzisnik C. Petrie G. Tereh B. Complete response of metastatic adrenal cortical carcinoma to o.p -DDD in a child. Pediatr Hematol Oncol. 1988:5:65-69.
84. Terzolo M. Daffara F. Ardito A. Zaggia B. Basile V. Ferrari L. Berruti A. Management of adrenal cancer: a 2013 update. Endocrinol Invest. 2014:37/31:207-17. doi: 10.1007/s40618-013-0049
85. Huang H. FoioT. Adjuvant mitotane for adrenocortical cancer - a recurring controversy. Journal of Clinical Endocrinology and Metabolism. 2008: 951101: 3730-3732. doi: 10.1210/ic.2008-0579
86. Terzolo M. Fassnacht M. Ciccone G. Allolio B. Berruti A. Adjuvant Mitotane for Adrenocortical Cancer—Working through Uncertainty. The Journal of Clinical Endocrinology & Metabolism. 2009: 94161:1879-1880. doi:10.1210/ic.2009-0120
87. Kroiss M. Ouinkler M. Eutz WK. Allolio B. Fassnacht M. Drug interactions with mitotane hv induction of CYP5A4 metabolism in the clinical management of adrenocortical carcinoma. Clinical Endocrinology: 2011:75:585-591. doi :10.1111 /i.1565-2265.2011.04214.x
88. Cordon-Cardo C. Q"Brien TP. Boccia T et al. Expression of the multidrug resistance gene product (T-glycoprotein) in human normal and tumor tissues. T Histochem Cvtochenr. 1990:38:1277-1287.
89. Flvnn SD. Murren TR. Kirbv WM et al. P-glvcoprotein expression and multidrug resistance in adrenocortical carcinoma. Surgery. 1992:112:981-986.
90. Goldstein LE Galski H. Foio A. et al. Expression of a multidrug resistance gene in human cancers. 1 Natl Cancer Inst. 1989:81:116-124.
91. Fridborg H. Larsson R. Tuhlin C. Rastad E Akerstrom G. Backlin K. Nvgren P P-glvcoprotein expression and activity of resistance modifying agents in primary cultures of human renal and adrenocortical carcinoma cells. An-ticancer Res. 1994:14:1009-1016.
92. Haak HR. van Seters AP. Moolenaar AT. Fleuren GT Expression of P-glvcoprotein in relation to clinical manifestation, treatment and prognosis of adrenocortical cancer. Eur T Cancer. 1993:29A: 1036-1038.
93. Fassnacht M. Terzolo M. Allolio В et al. Combination chemotherapy in advanced adrenocortical carcinoma. FIRM-ACT Study GroupN Engl T Med. 2012:366C23'):2189.
94. Sperone P. Ferrero A. Daffara F. et al. Gemcitabine plus metronomic 5-fluorouracil or capecitabine as a second-/third-line chemotherapy in advanced adrenocortical carcinoma: a multicenter phase II study. Endocr Relat Cancer. 2010:17('2):445-55.
95. Berruti A. Grisanti S. Pulzer A. et al. Long-term outcomes of adjuvant mitotane therapy in patients with radically resected adrenocortical carcinoma. Journal of Clinical Endocrinology and Metabolism.2017:102:1358-1365. doi:10.1210/ic.2016-2894
96. Fassnacht M. Hahner S. Polat B. et al. Effcacv of adjuvant radiotherapy of the tumor bed on local recurrence of adrenocortical carcinoma. Journal of Clinical Endocrinology and Metabolism. 2006:91:4501-4504.
97. Habra MA. Eiaz S. Feng L. et al. A Retrospective Cohort Analysis of the Effcacv of Adjuvant Radiotherapy after Primary Surgical Resection in Patients with Adrenocortical Carcinoma. Journal of Clinical F.ndocrinology and Metabolism. 2013:98:192-197. doi :10.1210/ic.2012-23671
98. Saholcb A. F.lse T. Grifftb KA. et al. Adjuvant radiation tberapv improves local control after surgical resection in patients with localized adrenocortical carcinoma. International Journal of Radiation Oncology. Biology. Physics. 2015:92:252-259. doi:10.1016/i.iirobp.2015.01.007
99. Cerquetti L. Bucci B. Marchese R. et al. Mitotane increases the radiotherapy inhibitory effect and induces G2-arrest in combined treatment on both H295R and SW13 adrenocortical cell lines. Endocrine-Related Cancer. 2008:15:623-634. doi:10.1677/erc.l.l315
100. Cerquetti L. Sampaoli C. Anrendola D. et al. Mitotane sensitizes adrenocortical cancer cells to ionizing radiations bv involvement of the cvclin Bl/CDK complex in G2 arrest and mismatch repair enzymes modulation. International Journal of Oncology. 2010:37:493-501.
101. Ahiven-Eepage G. Coste T. Tissier F. et al. Adrenocortical carcinoma and pregnancy: clinical and biological features and prognosis. European Journal of Endocrinology 2010:165:795-800. doi: 10.1550/ETE-10-04121
102. Sirianni R. Zolea F. Chinrento A. et al. Targeting estrogen receptor-alpha reduces adrenocortical cancer (ACC) cell growth in vitro and in vivo: potential therapeutic role of selective estrogen receptor modulators ISERMsl for ACC treatment, fournal of Clinical Endocrinology and Metabolism. 2012:97:E2238-E2250. doi:10.1210/ic.2012-2374
103. Icard P. Goudet P. Charpenav C. Adrenocortical carcinomas: surgical trends and results of a 253-patient series from the French Association of Endocrine Surgeons Study Group. World 1 Surg. 2001:25:891-897.
104. Cmcitti F. Bellantone R. Ferrante A. et al. Tbe Italian Registry for Adrenal Cortical Carcinoma: analysis of a multlinstitutional series of 129 patients. Tbe ACC Italian Registry Study Group. Surgery. 1996:119:161-170.
105. Pomniier RF. Brennan MF. An eleven-vear experience with adrenocortical carcinoma. Surgery 1992:112:963-970: discussion. 970-971.
106. Soreide 1A. Brabrand K. Thoresen SO. Adrenal cortical carcinoma in Norway. 1970-1984. World 1 Surg. 1992:16:663-667: discussion. 668.
107. Vassilopoulou-Sellin R. Schultz PN. Adrenocortical carcinoma. Clinical outcome at the end of the 20th century. Cancer. 2001:92:1113-1121.
108. lohanssen S. Hahner S. Saeger W. et al. Defcits in the management of patients with adrenocortical carcinoma in germanv. Deutsches Arztehlatt International.2010:107:11885-11889.
109. Bilinioria KY. Shen WT. F.larai D. et al. Adrenocortical carcinoma in the United States: treatment utilization and prognostic factors. Cancer. 2008:115:5150-5156. Idoi:10.1002/cncr.23886
110. Libe R. Borget I. Ronchi CL. et al. Prognostic factors in stage III-IV adrenocortical carcinomas (ACC): an European Network for the Study of Adrenal Tumor JEN SAT) study. Annals of Oncology.
111. Miller BS. Gauger PG. Hammer GD. Giordano T1 & DohertvGM. Proposal for nrodifcation of the ENSAT staging system for adrenocortical carcinoma using tumor grade. Langenbecks Archives of Surgery 2010 595 955-961.
112. Weiss EM. Medeiros ET. Vickery AE Tr. Pathologic features of prognostic signifcance in adrenocortical carcinoma. American Journal of Surgical Pathology. 1989:15:202-206. doi :10.1097/00000478-198905000-00004Morimoto R. Satoh F. Murakami O. et al. Inmrunohistochemistrv of aproliferation marker Ki67/MIB1 in adrenocortical carcinomas: Ki67/MIB1 labeling index is a predictor for recurrence of adrenocortical carcinomas. Endocrine Tournal.2008:55:49-55.doi: org/10.1507/endocri.K07-079
113. Asare EA. Wang TS. Winchester DP. et al. A novel staging system for adrenocortical carcinoma better predicts survival in patients with stage I/II disease. Surgery. 2014:156:1378-1386. doi:10.1016/i .surg. 2014.08.018
114. Abiven-Lepage G. Coste 1. Tissier F. et al. Adrenocortical carcinoma and pregnancy: clinical and biological features and prognosis. European Tournal of Endocrinology. 2010:163:
793-800. doi:10.1530/ElE-10-0412
115. De Corbiere P. Ritzel K. Cazabat L. et al. Pregnancy in women previously treated for an adrenocortical carcinoma. Journal of Clinical Endocrinology and Metabolism. 2015:100: 4604-4611. doi:10.1210/ic.2015-2341
116. Всероссийское общество редких (орфанных! заболеваний
118. Hough. A.i. Prognostic factors in adrenal cortical tumors. A mathematical analysis of clinical and morphologic data / A.l. Hough. l.W. Hollifield. D.h. Page et al. // Am 1 Clin Pathol. - 1979. -Vol. 72. - P. 390-399.
119. Aubert. S. Weiss system revisited: a clinicopathologic and immunohistochemical study of 49 adrenocortical tumors / S. Aubert. A. Wacrenier. X. Lerov et al. // Anr. 1. Surg. Pathol. - 2002. -Vol. 26. №12. - P. 1612-1619.
120. Papotti. M. The Weiss score and bevond - histopathologv for adrenocortical carcinoma / M. Papotti. R. Libe. E.Duregon et al. // Hornr. Cancer. - 2011. - Vol. 2. №6. - P. 333-340.
121. Volante. M. Pathological and molecular features of adrenocortical carcinoma: an update / M. Volante. C. Buttigliero. E. Greco et al. //1. Clin. Pathol. - 2008. - Vol. 61- №7. - P. 787-793.
122. Sasano. H. Recent advances in histopathologv and immunohistochemistry of adrenocortical carcinoma / H. Sasano. T. Suzuki. T. Moriva // F.ndocr. Pathol. - 2006. - Vol. 17. №4. - P. 345-354.
123. Beuschlein. F. Major Prognostic Role of Ki67 in Localized Adrenocortical Carcinoma After Complete Resection / F. Beuschlein. 1. Weigel. W. Saeger et al. //1 Clin Endocrinol Metab. 2015. -Vol. 100.-P.841-849.
124. Papathonras. T. An International Ki67 Reproducibility Study in Adrenal Cortical Carcinoma / T. Papathonras. E. Pucci. T. Giordano et al. //Anri Surg Pathol. - 2016. - Vol. 40. - P. 569-576.
125. Adrenal Cortical Carcinoma. In: Brierlev ID. Gospodarowicz MK. Wittekind C feds'). TNM Classification of Malignant Tumours 18th edition). Oxford. UK: Wilev-Blackwell. 2017.
126. Libe. R. Prognostic factors in stage III-IY adrenocortical cancinoma (ACC): an European Network for the study of adrenal tumor IF, NS ATI study / R. Libe. LBorget. L.Ronchi // Annals of Oncology.-2015.-Vol. 26.-P. 2119-2125.
127. Бохян. В.Ю.. Павловская А.И.. Губина Г.И.. Стилили И. С. Клиническая оттенка гистологических систем диагностики алренокортикалънътх опухолей // Архив патологии. - 2015. - №3. - Т. 77. - Стр. 17-22.
128. Стилиди И.С.. В.Ю. Бохян. И.Е. Карманов. Поражение нижней полой вены при адренокортикальном раке: результаты хирургического лечения // Анналы хирургии. - 2016. №4. - Т.21. - Стр. 248-256.
129. Fassnacht М. Kenn W & Allolio В. Adrenal tumors: How to establish malignancy? 1 Endocrinol Invest 2270 2004 27 387-399.
130. Legro RS. Arslanian SA. Ehrmann DA. Hoeger KM. Murad MH. Pasquali R & Welt CK. Diagnosis and 2279 treatment of polycystic ovary syndrome: an F.ndocrine Society clinical practice guideline. I Clin 2280 F.ndocrinol Metah 2013 98 4565-4592.
131. ChortisV.TavlorAF.SchneiderP.TomlinsonlW.HughesBA.O’NeilDM.biheR.AllolioB.BertagnaX.BertheratL et al. Mitotane therapy in adrenocortical cancer induces CYP3A4 and inhibits 5a-reductase. explaining the need for personalized glucocorticoid and androgen replacement. Journal of Clinical Endocrinology and Metabolism 2013 98 161-171.
132. Rodriguez-Galindo C. Figueiredo BC. Zambetti GP. et al.: Biology, clinical characteristics, and management of adrenocortical tumors in children. Pediatr Blood Cancer 45 131: 265-73.2005.
133. Zancanella P. Pianovski MA. Oliveira BH. et al.: Mitotane associated with cisplatin. etoposide. and doxorubicin in advanced childhood adrenocortical carcinoma: mitotane monitoring and tumor regression. I Pediatr Hematol Oncol 28 (81: 513-24. 2006.
134. Berruti A. Terzolo M. Pia A. et al. Mitotane associated with etoposide. doxorubicin, and cisplatin in the treatment of advanced adrenocortical carcinoma. Italian Group for the Study of Adrenal Cancer. Cancer. 1998:83:2194-2200
Приложение А1. Состав рабочей группы
Мельниченко Галина Афанасьевна, д.м.н., профессор, академик РАН, заместитель директора Центра - Директор Института клинической эндокринологии ФГБУ «НМИЦ эндокринологии» Минздрава РоссииАлексеев Борис Яковлевич, д.м.н., профессор, заместитель генерального директора по науке ФГБУ «НМИЦ радиологии» Минздрава России
Бельцевич Дмитрий Германович, д.м.н., профессор, главный научный сотрудник ФГБУ «НМИЦ эндокринологии» Минздрава России
Стилиди Иван Сократович, д.м.н., профессор, академик РАН, Директор ФГБУ «НМИЦ онкологии им. Н.Н. Блохина» Минздрава России
Горбунова Вера Андреевна, д.м.н., профессор, ведущий научный сотрудник ФГБУ «НМИЦ онкологии им. Н.Н. Блохина» Минздрава России
Кузнецов Николай Сергеевич, д.м.н., профессор, заведующий отделом хирургии ФГБУ «НМИЦ эндокринологии» Минздрава России
Бохян Ваган Юрикович, д.м.н., заведующий хирургическим отделением №2 (диагностики опухолей) ФГБУ «НМИЦ онкологии им. Н.Н. Блохина» Минздрава России
Коломейцева Алина Андреевна, к.м.н., заведующий дневным стационаром Поликлинического отделения МНИОИ им. П.А. Герцена - филиал ФГБУ «НМИЦ радиологии» Минздрава России
Жуков Николай Владимирович, к.м.н., доцент кафедры онкологии, гематологии и лучевой терапии ФГБОУ ВО РНИМУ им. Н.И. Пирогова Минздрава России, руководитель отдела оптимизации лечения подростков и молодежи с онкологическими заболеваниями ФГБУ «НМИЦ ДГОИ им. Дмитрия Рогачева» Минздрава России
Рослякова Анна Александровна, научный сотрудник ФГБУ «НМИЦ эндокринологии» Минздрава России
Невольских Алексей Алексеевич, д.м.н., заместитель директора по лечебной МРНЦ им. А.Ф. Цыба - филиал ФГБУ «НМИЦ радиологии» Минздрава России
Иванов Сергей Анатольевич, д.м.н., профессор РАН, директор МРНЦ им. А.Ф. Цыба -филиал ФГБУ «НМИЦ радиологии» Минздрава России
Хайлова Жанна Владимировна, к.м.н., заместитель директора по организационно-методической работе МРНЦ им. А.Ф. Цыба - филиал ФГБУ "НМИЦ радиологии" Минздрава России.
Геворкян Тигран Гагикович, заместитель директора НИИ КЭР ФГБУ ФГБУ «НМИЦ онкологии им. Н.Н. Блохина» Минздрава России
Конфликт интересов:
Авторы декларируют отсутствие конфликта интересов.
Приложение А2. Методология разработки клинических рекомендации
Предлагаемые клинические рекомендации, на настоящий момент, являются основным согласительным документаом, регулирующим положения практической деятельности врачей при АКР.
Настоящие Клинические рекомендации направлены на улучшение результатов диагностики и лечения пациентов с адренокортикальным раком.
Настоящие Клинические рекомендации не рассматривают абсолютно все аспекты проблемы и возможные исключения из правил.
Настоящие Клинические рекомендации не могут гарантировать определенный результат и при этом они не устанавливают стандарты ведения пациентов.
Настоящие Клинические рекомендации не предназначены для лечения конкретного пациента. Решение об оптимальной тактике ведения каждого пациента должны основываться на результатах обследования с учетом индивидуальных обстоятельств.
Редкость АКР обусловливает объективные трудности из-за отсутствия проспективных исследований достаточной мощности и достаточного опыта лечения этого заболевания вне специализированных учреждений, концентрирующих таких пациентов. За рубежом в течение последних 30-40 лет проведены крупные, как проспективные, так и ретроспективные исследования, часть из которых включают в себя до 500 пациентов. К сожалению, в России исследований с подобным объемом выборки не проводилось. В связи с этим, настоящие Клинические рекомендации в основном опираются на зарубежную доказательную базу и учитывают экспертное мнение зарубежных исследователей.
В основу клинических рекомендаций положены существующие консенсусы и клинические рекомендации: (1) Классификация Всемирной Организации Здравоохранения (WHO) опухолей эндокринной системы, 4-е издание, 2017 год; (2) Клинические рекомендации по диагностике и лечению адренокортикального рака Европейского общества эндокринологов (European Society of Endocrinology), совместно с Европейским обществом по изучению опухолей надпочечников (European Network for the Study of Adrenal Tumors), 2018 год; (3) Клинические рекомендации по диагностике и лечению адренокортикального рака, 2015 год - а также отечественные и зарубежные научные работы по данной проблематике.
Методы, использованные для сбора/селекции доказательств: проводился поиск в электронных базах данных по ключевым словам, связанным с АКР и соответствующими разделами клинических рекомендаций; проводилась оценка качества и релевантности найденных источников.
Описание методов, использованных для сбора/селекции доказательств:
доказательной базой для рекомендаций являются публикации, вошедшие в Кохрановскую библиотеку, базы данных EMBASE и MEDLINE, e-library. Елубина поиска составляла до 40 лет.
Методы, использованные для оценки уровней достоверности доказательств:
• Консенсус экспертов;
• Оценка значимости в соответствии с уровнями доказательности.
В связи с орфанностью заболевания и отсутствием проспективных исследований достаточной мощности ряд рекомендаций характеризуется низким уровнем доказательности по формальному признаку.


Методы, использованные для оценки уровней достоверности доказательств:
• Консенсус экспертов;
• Оценка значимости в соответствии с уровнями доказательности.
В связи с орфанностью заболевания и отсутствием проспективных исследований достаточной мощности ряд рекомендаций характеризуется низким уровнем доказательности по формальному признаку.
Диагностические и лечебные опции в отношении пациентов с АКР требуют мультидисциплинарного подхода.
Порядок обновления клинических рекомендаций
Механизм обновления клинических рекомендаций предусматривает их систематическую актуализацию - не реже чем один раз в три года, а также при появлении новых данных с позиции доказательной медицины по вопросам диагностики, лечения, профилактики и реабилитации конкретных заболеваний, наличии обоснованных дополнений/замечаний к ранее утверждённым КР, но не чаще 1 раза в 6 месяцев.
Приложение АЗ. Связанные документы
КР400. Хронический болевой синдром (ХБО у взрослых пациентов, нуждающихся вРекомендации по заместительной терапии при лечении митотаном
1. Абсолютная недостаточность глюкокортикоидов
• Глюкокортикоиды: с начала лечения митотаном’1"1' дозы превышают стандартные:
о Гидрокортизон’1"1' 50-75 мг/сут
• Оценка адекватности заместительной терапии производится по клиническим критериям, включая артериальное давление, пульс, наличие/отсутствие аппетита, динамику массы тела. Также необходимо принимать во внимание уровень электролитов крови.
2. Минералкортикоидная недостаточность регулярно оценивается по уровню электролитов крови, коррекция - флудрокортизон**
3. Гипогонадизм у мужчин оценивается по клиническим проявлениям (эректильная дисфункция и гинекомастия), подтверждается результатами гормонального обследования, требует заместительной терапии.
4. Вторичный гипотиреоз регулярно оценивается по уровню ТГГ, Т4 св. (ТГГ - норма, снижение св. Т4 - снижен) и требует заместительной терапии.
Рекомендации по применению митотана в качестве адъювантной терапии
• Начальная доза митотана’1"1' составляет 1 г/сут (2 табл/сут). Каждые 3-7 дней проводится повышение на 0.5 - 2 г/сут (с учетом переносимости) до дозы 4-8 г/сут (8-16 табл/сут), или до максимально переносимой дозы; препарат принимается 2-5 раз/сут во время еды.
• Повышение дозы регулируется
о переносимостью препарата;
о достигнутой терапевтической концентрацией (14-20 мкг/мл)
• Оценивать уровень митотана’1"1' в крови необходимо каждые 4-8 нед до достижения целевой зоны;
° при достижении целевой зоны каждые 2-3 мес в течение 2 лет, в дальнейшем возможна оценка через более длительные интервалы
• Клиническая и топическая оценка проводится не реже 1 раз в 2-3 месяца
• Необходимо определение уровня митотана’1"1' в крови при возникновении побочных эффектов
• При возникновении побочных эффектов:
о незначительных/умеренных - продолжить прием митотан, проводить симптоматическую терапию;
о значительных - вернутся к последней переносимой дозе, проводить симптоматическую терапию;
о тяжелых - прекратить прием митотана’1"1', провести специфическое лечение симптомов, затем возобновить митотан’1"1', начиная с малых доз.
Приложение Б. Алгоритмы ведения пациента




Приложение В. Информация для пациентов
Рак коры надпочечника, или адренокортикальный рак (АКР) - редкое (орфанное) опухолевое заболевание, характеризующееся, как правило, поздним сроком выявления и, при несвоевременном и неполном лечении, неблагоприятным прогнозом. АКР примерно в половине наблюдений может сопровождаться повышенным выделением в кровь гормонов надпочечников [115].АКР - одна из самых редких опухолей человека. Ежегодно выявляют 0,5-2 случая АКР на миллион населения. Женщины болеют в 2.5 раза чаще мужчин, средний возраст пациентов составляет 46 лет. В России АКР внесен в Перечень редких ('орфанных) заболеваний Минздрава РФ.
Причины развития АКР
Причины возникновения и прогрессирования АКР, очевидно, заключаются в генетических дефектах. Они могут затрагивать исключительно клетки коры надпочечника, из которых и развивается опухоль. В этом случае заболевание затрагивает только самого пациента и не наследуется его детьми. Гораздо реже (3-5% от всех случаев АКР среди взрослых пациентов) АКР развивается из-за врожденных генетических дефектов, присутствующих во всех клетках организма. В этом случае говорят о развитии АКР в рамках наследственного синдрома. На сегодняшний день, АКР описан как компонент следующих синдромов: Ли-Фраумени, Линча, множественной эндокринной неоплазии 1 типа, Гарднера (семейный аденоматозный полипоз), комплекса Карни, Беквита-Вайдемана, нейрофиброматоза 1 типа. В случае, если у пациента выявлен АКР в составе наследственного синдрома, необходимо обследовать его кровных родственников и, прежде всего, детей.
Важно отметить, что среди всех случаев АКР в детском возрасте 80% обусловлены врожденными генетическими дефектами, приводящими к развитию синдрома Ли-Фраумени.
Четкой связи между возникновением АКР и образом жизни или внешними факторами до настоящего времени не обнаружено.
Проявления АКР
В 50-60% случаев АКР сопровождается повышенной продукцией гормонов коры надпочечников, а именно:
1. глюкокортикоиды (кортизол и кортизон);
2. минералокортикоиды (альдостерон, кортикостерон и дезоксикортикостерон);
3. половые гормоны (андрогены и эстрогены).
Большинство опухолей изолированно секретирует кортизол (до 60%) или андрогены в комбинации с кортизолом (30%).
Жалобы пациентов с гормонально-активным АКР определяются повышенной продукцией соответствующих гормонов:
- кортизола (общая слабость, головные боли, ожирение, с типичным отложением жировой клетчатки в области лица, шеи, груди, живота. Лицо при этом выглядит округлым, лунообразным. На щеках появляется пурпурный румянец. Руки и ноги наоборот становятся тонкими из-за уменьшения массы мышц. На коже появляются угревые высыпания. Раны и порезы заживают медленно. Возникают боли в костях и склонность к переломам, бесплодие, повышение артериального давления и др.);
- альдостерона (повышение артериального давления, слабость мышц вследствие потери калия);
- тестостерона (рост нежелательных волос на теле, понижение тембра голоса, увеличение клитора у женщин, повышение жирности и нечистота кожи);
- эстрогенов (импотенция, увеличение грудных желез у мужчин, маточные кровотечения у женщин в постменопаузе).
Гормонально-неактивные опухоли встречаются в 40% случаев и коварны потому, что долгое время могут никак о себе не заявлять и в дальнейшем, при увеличении размеров опухоли, проявляться общими неспецифичными симптомами: дискомфортом или болями в животе или спине, ощущением переполнения в животе, наличием объемного образования в брюшной полости, определяемого врачом или самостоятельно пациентом при ощупывании живота.
Выявление (диагностика) АКР
Широкое внедрение в клиническую практику методов лучевой диагностики, таких как ультразвуковое исследование (УЗИ), компьютерная томография (КТ), магнитно-резонансная томография (МРТ), привело к резкому увеличению числа случайно выявленных опухолей надпочечников. Случайно выявленная опухоль надпочечника может оказаться как гормонально-неактивной, так и активно производить различные гормоны (смотри выше); исходить из различных частей надпочечника, быть злокачественной или доброкачественной. Среди всех случайно выявленных опухолей надпочечника АКР встречается примерно в 4% случаев.
При подозрении на АКР проводится лабораторная диагностика, которая заключается в выявлении гормональных нарушений, характерных для гормонально-активного АКР. Для подтверждения/исключения гормональной активности опухоли рекомендованы следующие методы лабораторной диагностики:
• определение кортизола в ранние утренние часы на фоне подавляющего теста с дексаметазоном;
• при отсутствии физиологического подавления уровня кортизола - определение адренокортикотропного гормона в утренние часы;
• всем пациентам с выявленной опухолью надпочечника для исключения другой опасной опухоли - феохромоцитомы, показано определение норметанефрина и метанефрина в суточной моче или плазме;
• при наличии у пациента с опухолью надпочечника артериальной гипертензии показано определение соотношения между уровнем альдостерона и активностью ренина плазмы для исключения первичного гиперальдостеронизма;
• при подозрении на изолированную или сочетанную (с гиперкортицизмом) опухолевую гиперпродукцию половых гормонов показано определение стероидных гормонов сыворотки крови (дегидроэпиандростерон-сульфат, 17-оксипрогестерон, андростендион, тестостерон, 17-р-эстрадиол у мужчин и женщин в менопаузе).
Методы лучевой диагностики на современном этапе имеют важнейшее значение для дооперационного подтверждения диагноза АКР. И если наличие прямых признаков злокачественности опухоли, таких как прорастание в окружающие органы, метастатическое поражение лимфоузлов, печени, легких, делает диагноз АКР практически очевидным, то их отсутствие ставит задачу по выявлению признаков, характерных именно для этой опухоли.
Компьютерная томография (КТ) - «золотой стандарт» лучевой диагностики опухолей надпочечников.
Ультразвуковое исследование (УЗИ) используется довольно часто в качестве первичного диагностического метода. УЗИ очень хорошо определяет опухоль, но, к сожалению, не всегда может точно определить происхождение этой опухоли. УЗИ может быть использовано для первичной диагностики опухолей надпочечников в случае невозможности выполнения компьютерной томографии.
Магнитно-резонансная томография (МРТ) рекомендована для первичной диагностики опухолей надпочечников, а также для исключения метастазов в головном мозге. МРТ обладает высокой чувствительностью в выявлении опухолей надпочечников, оценки состояния опухоли, в том числе поражения соседних органов. Однако точность данного метода пока ниже, чем КТ.
Остеосцинтиграфия (сцинтиграфия костей) рекомендована при подозрении на метастазы костей скелета.
Позитронно-эмиссионная томография (ПЭТ) с дезоксифторглюкозой (ФДГ) показана для определения стадии и распространенности процесса, а также в качестве диагностического метода, позволяющего определить злокачественный потенциал при небольших размерах опухоли (до 4 см).
Пункционная биопсия в случае АКР не имеет доказанных преимуществ, обладает низкой чувствительностью, недостаточной информативностью и высокой вероятностью осложнений, поэтому ее проведение не показано.
Лечение АКР
Диагностика и лечение АКР должны происходить с участием врача-эндокринолога, врача-хирурга, врача-онколога, врача-клинической лабораторной диагностики, врача-радиолога в специализированном учреждении/отделении.
Наиболее эффективным методом лечения является хирургический. При выявлении АКР на ранних стадиях заболевания, когда нет прорастания опухоли в жизненно важные органы и/ или многочисленных метастазов, операция является первым этапом комплексной терапии. Далее при отсутствии метастазов хирургическое лечение может быть дополнено назначением ЛС в адъювантном режиме). Решение о лечении принимается на основании особенностей опухоли (прежде всего, индекс Ki67) и течения АКР у данного пациента.
В случае наличия метастазов на момент выявления АКР или возникновения их в дальнейшем комплексное лечение должно включать и химиотерапию.
Множественные метастазы и/или прорастание опухоли в жизненно важные органы на момент постановки диагноза АКР, как правило, не позволяют провести адекватное хирургическое лечение или могут привести к серьезным осложнениям во время операции. Поэтому в такой ситуации лечение начинают с химиотерапии, что, в большинстве случае, приводит к уменьшению размеров опухолевых очагов; далее проводят операцию.
Дистанционная лучевая терапия (ДЛТ) рекомендована в качестве паллиативной терапии при метастазах АКР в кости и центральную нервную систему.
Таргетная терапия в настоящее время не рекомендована для лечения пациентов с распространенным АКР, т.к. в ряде исследований возможности применения препаратов этого класса при распространенных формах АКР были продемонстрированы неудовлетворительные результаты.
Прогноз
В целом, прогноз АКР определяется своевременностью выявления заболевания, эффективностью проводимого лечения и приверженностью пациента к терапии.
Лечение АКР является длительным и трудоемким процессом, требует усилий не только команды врачей и самого пациента. Приверженность к лечению, выполнение рекомендаций по своевременному обследованию и лечению, как правило, позволяют добиться улучшения или стабилизация АКР. Наиболее неблагоприятный прогноз отмечается у пациентов, которые отрицают свое заболевание и не выполняют рекомендаций лечащего врача.
Приложение Г.
Приложение Г1. Критерии оценки ответа опухоли на лечение (RECIST 1-1)
Название на русском языке: Критерии оценки ответа опухоли на химиотерапевтическое лечение (RECIST 1.1)
Оригинальное название: Response evaluation criteria in solid tumors 1.1 (RECIST 1.1) Источник (публикация с валидацией):
Eisenhauer ЕА, Therasse Р, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M et al: New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009,45(2):228-247
Источник (на русском языке):
Еомболевский ВА, Лайпан АШ, Шапиев АН, Владзимирский АВ, Морозов СП (ред.): Методические рекомендации по применению критериев ответа солидных опухолей на химиотерапевтическое лечение (RECIST 1.1). Методические рекомендации № 46; 2018. http ://nredradiology.moscow/d/l 364488/d/no46_2018_recist_l 1 .pdf
Тип: Шкала оценки
Назначение: оценка ответа на химиотерапевтическое лечение


Приложение Г2. Критерии адекватности операции
Название на русском языке: Классификаций резидуальных опухолей (R Классификация) Оригинальное название: Residual Tumour (R) Classification Источник (публикация с валидацией):
Brierley J, Gospodarowicz МК, Wittekind Ch (eds). TNM classification of malignant tumours. Oxford, UK ; Hoboken, NJ: John Wiley & Sons, Inc., 2017.
Тип: Шкала оценки
Назначение: оценка адекватности хирургического лечения по наличию/отсутствию резидуальной опухоли

Теги: рак
234567 Начало активности (дата): 23.12.2022 22:32:00
234567 Кем создан (ID): 989
234567 Ключевые слова: надпочечник, рак, первичная опухоль
12354567899
Похожие статьи
Рак носоглоткиКурс медицинской рентгенологии. Специальная рентгенотерапия. Часть 3. Глава 15.5
Рентген на дому 8 495 22 555 6 8
Курс медицинской рентгенологии. Рентгенотерапия.Часть 3. Глава 14.4
Курс медицинской рентгенологии. Глава 1


