 11.08.2024
11.08.2024
Механизм наследования идиопатического сколиоза
Этиологический фактор идиопатического сколиоза (ИС) является предметом дискуссий ученых многих профилей.
Сколиотическая болезнь известна человечеству с древнейших времен. С тех пор делаются попытки объяснить причины возникновения этой тяжелой патологии, инвалидизирующей детей и подростков. Проходили столетия, возникали новые теории, развивались старые, на основе которых предлагались методы лечения, но по-прежнему сколиотическая болезнь оставалась «крестом ортопедии». Канули в лету рахитическая, остеопатическая теории, школьный сколиоз, но все еще обсуждаются весьма сомнительные с точки зрения обоснованности гормональная и биомеханическая теории. В последние годы наиболее популярными стали исследования, касающиеся наследственной природы заболевания.
Основанием для подобных работ явились наблюдения семейного характера идиопатического сколиоза (ИС) [2]. В этом аспекте сформировались три направления:
Исследования типа и характера наследования ИС.
Попытки идентифицировать ген и (или) гены, ответственные за развитие ИС.
Моделирование генетически зависимых деформаций позвоночника. В различных работах предлагались разные модели наследования ИС: мультифакторная [32], модель сцепленного с полом наследования [13] и модель майоргенного наследования болезни с неполной пенетрантностью генотипов [2, 9]. Обсуждалась возможность генетической гетерогенности ИС.
Так, в работе [9] было показано, что распределение заболевания среди родственников 241 пробанда можно объяснить как наличием аутосомного майоргена с неполной пенетрантностью генотипов, так и генетически гетерогенным контролем: в одних семьях распределение заболевания описывается доминантной моделью, в других - мультифакторной. Противоречивость данных разных исследователей может быть объяснена некорректной выборкой родословных, малым набором тестируемых гипотез и т. д.
Попытка идентифицировать гены, ответственные за развитие ИС, является прерогативой последних десятилетий. В качестве претендентов рассматривались структурные гены внеклеточного матрикса: эластина коллагена, фибриллина [10, 23]. Однако ни один из этих генов не продемонстрировал причастность к развитию болезни. В связи с тем, что формирование деформации позвоночника при ИС происходит в период полового созревания и активного роста [20], логичными были исследования связи полиморфизма гена-рецептора и эстрогена, но и этот вариант не принес желаемого результата.
Противоречивые результаты получены и при сканировании генома [26]. В одной большой семье была обнаружена связь с участком хромосомы 10q [31]. Исследования семи семей из китайской популяции [11], в которой 25 человек страдали ИС, выявили связь возникновения болезни с 19-й хромосомой. Попытки увязать этиологию ИС с нарушениями в экспрессии локусов Х-хромосомы также не увенчались успехом [21].
В 1977 г. был обнаружен полиморфизм гена-агрекана человека, обусловленный различным числом повторов в экзоне G3 [9]. Этот экзон кодирует домен, к которому присоединяются цепи хондроитинсульфата. Размер этого домена, а следовательно, и количество присоединенных цепей зависит от числа тандемных повторов. Изменение количества хондроитинсулфата оказывает влияние на физико-химические свойства агрекана и функциональные свойства хрящевой ткани [9]. Некоторыми исследователями уже были предприняты попытки использовать этот полиморфизм в изучении известных патологий хрящевой ткани.
В 1998 г. Хортон и др. обнаружили ассоциацию между одним аллелем агрекана (с числом повторов 27) и двусторонним остеоартритом суставов кисти. Однако для двустороннего остеоартрита коленного сустава таких ассоциаций найдено не было [19]. В 1999 г. японскими учеными Кавагучи и др. [22] было показано, что множественная и наиболее ярко выраженная дегенерация поясничных межпозвонковых дисков коррелирует с присутствием аллелей агрекана, характеризующихся малым числом тандемных повторов. Это подтверждает возможность влияния длины G3 экзона на физические свойства хряща пластинки роста (ПР) тела позвонка.
Весьма привлекательным является создание моделей деформаций позвоночника, фенотипически подобных ИС человека, на животных. Исследователи путем ферментативного импринтинга и последующего генетического анализа вывели линию крыс с генетически детерминированной деформацией позвоночника [4]. Наиболее многочисленны моделирования деформаций позвоночника на мышах. Описано более 250 подобных мутаций у мышей с кифотическими и сколиотическими деформациями позвоночника [16].
Значительный интерес представляет тот факт, что 28 подобных мутаций локализовано в генах, имеющих гомологию с генами человека.
Однако работ, касающихся проверки генов-кандидатов у человека, в доступной литературе нет. Надо полагать, что такие исследования ждут своих авторов.
Таким образом, механизмы наследования ИС до сих пор не установлены. В связи с этим целью настоящего исследования явилось следующее: на репрезентативных выборках родословных исследовать механизм генетической детерминации ИС и определить наследственно-фенотипические признаки данной патологии.
Материал и методы
Метод генетического обследования включал в себя составление родословных и их сегрегационный анализ. Информация для составления родословных собиралась путем очного опроса пробандов и записывалась по общепринятой схеме.
Для изучения пусковых механизмов развития ИС клинико-генетическими методами обследовано 92 пробанда и 919 членов семей, на основе которых были сформированы три группы:
Дети (пробанды) с верифицированным диагнозом ИС, лечившиеся в специальной школе-интернате для больных сколиозом. Эта группа включала 92 пробанда в возрасте от 7 до 17 лет. Среди них было 17 мальчиков (18 %) и 75 девочек (82 %). Соотношение полов соответствовало литературным данным [32, 33]. Сколиоз I степени выявлен у 23 % девочек, II - у 47 %, III-IV - у 30 %.
2. Родственники больных ИС I-III степени родства (719 человек) в возрасте 21-60 лет. Обследовано 92 матери, из них сколиозом не поражены (без признаков сколиотической деформации или с деформацией по Cobb менее 10°), что составило 55 %, пораженные сколиозом - 41 (45 %). Отцов обследовано 88, пораженных сколиозом - 32 (36 %), не пораженных - 56 (64 %). Сибсов обследовано 62, из них больных сколиозом - 31 (50 %) и здоровых - 31 (50%).
3. Родословные семей с ИС. Эта группа состояла из 92 человек. В каждой родословной присутствовал только один пробанд. Родословные различались по своей структуре. Так, 41 родословная была представлена только парой родителей и их детьми (ядерная родословная), 11 из этих родословных имели только одного потомка, 27 - двух, трех и более потомков;
родословных состояли из трех или четырех поколений. Из всех обследованных семей в 29 (32 %) все родственники I степени родства были здоровы, в 17 (18,4 %) сколиозом поражены только матери, в 12 (13 %) - только отцы, в 4 (5,4 %) только сибсы, в 4 (4,3 %) сколиозом страдали и отец, и мать, в 11 (11,9 %) - мать и сибс, в 5 (5,4 %) - отец и сибс, в 9 (9,7 %) - все родственники I степени родства.
Средний размер родословных составил 11 человек. Получена информация о фенотипах 719 особей. Все пробанды и родственники подвергались клиническому обследованию, включающему рентгенографию и компьютерно-оптическую топографию. В ходе обследования фиксировались следующие параметры: степень сколиотической деформации [12], торсия тел позвонков [29], локализация и сторона деформации. С помощью компьютерно-оптической топографии проводилось описание поверхности спины, торсии тел позвонков и рассчитывались основные параметры деформации позвоночника. Степень деформации определялись по классификации В.Д. Чаклина.
Сегрегационный анализ родословных проводился с помощью специально разработанной версии программы МАН-2 [1], позволяющей рассматривать три компонента - майор- генный, полигенный и средовой. Данная программа предназначена для проверки моногенных диаллельных гипотез о наследовании альтернативного признака по выборкам из популяции родословных произвольной структуры, сделанных по пробандам. Она позволяет проводить анализ альтернативных признаков с неполной пенетрантностью генотипов, зависящей от различных характеристик (пол, возраст и т. д.).
Наследование признака описывается в рамках моногенной диаллельной модели с использованием следующих параметров:
g (g = A1A1 А1А2 или A2A2) - генотип;
q - частота вызывающего болезнь аллеля А2;
w (g) - пенетрантность генотипа g (g = A1A1, А1А2 или A2A2), то есть вероятность развития болезни у особи с генотипом g;
T и а - параметры возрастной зависимости;
T(g) - переходная вероятность или вероятность того, что особь с генотипом g передаст потомку аллель А1. В случае менделевской сегрегации генов переходные вероятности равны 1; 0,5 и 0 для g = A1A1, А1А2 или A2A2 соответственно.
Оценки параметров в этой программе получают методом максимума правдоподобия.
При тестировании гипотез используется критерий отношения правдоподобия [24] (функции правдоподобия LH), то есть вероятность родословных, выраженных через параметры генетической модели. Для тестирования менделевской сегрегации генов используется предложенный Эльстоном и Стюартом [14] вариант критерия отношения правдоподобия 2(lH2-LH1) = х2. Он основан на сравнении с тремя гипотезами: с менделевскими, произвольными и равными переходными вероятностями:
H : Т1 = 1. Т2 = 0.5; Т3 = 0;
Н2 : Tg;
Н3 : Т1 = Т2 = Т3 Если первые две из этих гипотез достоверно не отличаются друг от друга (х2 < 7,81), а третья значимо хуже второй (х2 < 5,99) менделевская сегрегация майоргена считается доказанной [15].
Среди различных математико-статистических методов обработки базы данных были выбраны следующие:
критерий Pearson х2 и максимальная вероятность (M-L), х2 для характеристики распределения качественных признаков в выбранной системе оценок;
корреляционно-регрессионный анализ наличия взаимной сопряженности между количественными признаками (топографические показатели).
Уровень достоверности принят 5 %. Статистический анализ полученных данных выполнен с использованием стандартного пакета программ «STATISTICA for Windows» для персональных компьютеров и с использованием специализированных руководств.
Из данных, которые ранее использовались для сегрегационного анализа [3, 8], были отобраны 33 пробанда с выраженными формами сколиоза II-IV степени и их ближайшие родственники. Число тандемных повторов в экзоне G3 гена-агрекана было определено у 115 человек. Среди них информативными оказались 31 тройка (больной ребенок и оба родителя) и 3 пары (больной ребенок и один из родителей). Выделение геномной ДНК из ядросодержащих клеток крови пациентов проводили с использованием фенола и хлороформа согласно протоколу FBI. ДНК экзона была получена методом ПЦР Реакцию проводили в следующих условиях: 67 мМ трис-HCl, рН 8,9; 16 мМ ^N4)^4; 1.5 мМ MgCl2; 0,01 % твин-20; 10мМ Р-меркапно-этанол; 80 мкМ каждого dNTP; по 30 нг олигонуклеиновых праймеров AGCl и AGC2; 50-150 нг геномной ДНК человека; 0,5 ед. Taq ДНК полимеразы.
Праймеры AGC1 - 5’- TAGAG-GCCCTA-CCGCA-GAGGT-AGAA-3’ (Тпл = 62 °С) были синтезированы В.Ф. Кобзевым (ИЦиГ СО РАН). Условия амплификации были следующими: 38 циклов, плавление - 94 °С/мин, полимеризация - 73 °С/мин. Продукты амплификации разделяли с помощью электрофореза в 2 % агарозном геле в присутствии бромистого эти- дия. Бэнды, соответствующие ампли- фикатам разного размера, были четко различимы на трансиллюминаторе.
Тестирование аллельных ассоциаций проводили методом TDT (transmission disequilibrium test) [28]. Материалом для этого метода являются больные потомки и их родители.
Анализируются распределения родительских аллелей, переданных и не переданных больному потомку. Эти данные полиморфного маркера, имеющего к аллелей, представляются в виде матрицы кх к, где в каждой ячейке расположено число родителей, передавших больному потомку аллель i и не передавших аллель j (nip. Статистика в асимптотике имеет распределение хи-квадрат с числом степеней свободы к-1. Здесь ni - суммирование по i-му столбцу.
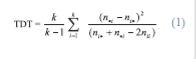
Эта форма TDT позволяет оценить общую неравновесность по сцеплению. Чтобы оценить ассоциацию болезни с конкретным аллелем, делается следующее: выбирается один из аллелей анализируемого локуса, а все остальные объединяются в общую группу [27]. В этом случае данные можно представить в виде матрицы 2 х 2. Тогда TDT-статистика, определяемая, как
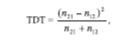
имеет в асимптотике распределение хи-квадрат с одной степенью свободы.
Результаты
При анализе клинико-генетических характеристик детей и родителей с ИС были выделены качественные и количественные параметры, которые оказались значимыми для оценки наследуемости признаков и характера течения болезни. Несомненный интерес представляет выявленная статистически достоверная связь (р = 0,02) наличия ИС у детей и родителей. Более того, экспрессивность признака нарастала в зависимости от тяжести деформации у детей (табл. 1).
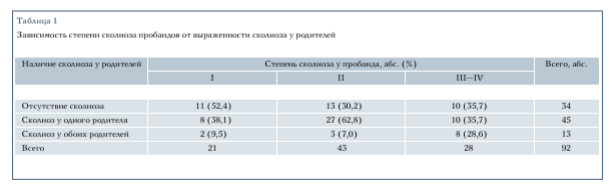
Так, в группе пробандов с I степенью ИС только 9,5 % имели больных родителей, а в 52,4 % родители были здоровы. Эти данные подтверждают гетерогенность группы детей с I степень ИС.
Среди обследованных детей III-IV степень ИС регистрировали у дево- чек-пробандов - 34,7 %, у мальчиков - 11,8%. Подобную зависимость можно объяснить более ранним интенсивным ростом девочек и превалированием у них более тяжелых грудных деформаций позвоночника (40 % у девочек и 29,4 % у мальчиков).
Попытка объяснить развитие лево-, правосторонних деформаций с позиции генетически обусловленной латеризации центральной нервной системы (ЦНС), индуцирующей развитие асимметрии формы [17], не получила подтверждения. Более убедительным представляется объяснение особенностями нарушения генетической регуляции разных зон тел позвонков в онтогенезе [6]. На основании ретроспективного анализа- наблюдения (в течение четырех лет) за 500 детьми, лечившимися в школе- интернате № 133 Новосибирска, созданы прогностические параметры закономерностей прогрессирования ИС (табл. 2).
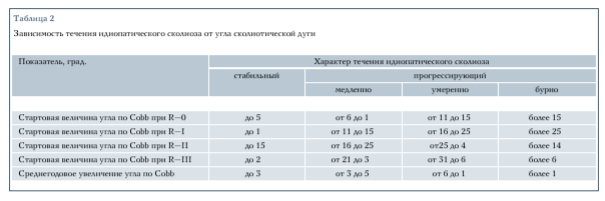
Анализ зависимости течения ИС у пробандов от частоты и степени ИС у родителей (преимущественно матерей) показал, что среди пробандов с бурным прогрессированием деформации 60 % родителей имели ИС, а среди пробандов со стабильным течением в 71,4 % родители были здоровы и только у 28,6 % родителей наблюдалась деформация позвоночника I-II степени. Кроме того, наблюдалось совпадение наследования в ассоциации «пробанд - мать» (р = 0,04). Подобной ассоциации «пробанд - отец» не наблюдалось (р = 0,78).
Весьма показательным является отсутствие в 76,9 % случаев деформации позвоночника у сибсов при стабильном течении болезни у пробанда, у 15 % сибсы имели сколиоз I степени, только у 7,7 % был диагностирован сколиоз II степени. Сколиоз III и IV степени у пробандов этой группы не был выявлен. В случае прогрессирующего течения сколиоза у пробанда число сибсов с выраженными формами сколиоза составляло 30,5 %. Анализ полученных данных поставил вопрос о диагностических критериях ИС. Сначала следует понять, являются ли медленно прогрессирующие деформации сколиотической болезнью (истинным сколиозом по нашей терминологии). На основании клиникогенетических исследований эти деформации позвоночника следует рассматривать как дисбаланс роста, то есть временное физиологическое несоответствие темпов роста мышечной и костной систем. Локализация сколиотической деформации, наличие ИС в семье являются важными прогностическими признаками, указывающими на возможность прогрессирования патологии у детей с ИС.
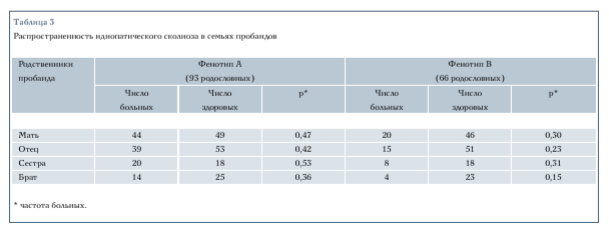
В табл. 3 показана частота сколиоза у различных родственников про- бандов. Видно, что во всех группах
родственников частота ИС значительно выше, чем в среднем в популяции. Это подтверждают известные данные о семейной агрегации изучаемой патологии. Кроме того, видно, что при любом задании дихотомического фенотипа частота сколиоза среди сестер пробандов выше, чем среди братьев. Это также хорошо согласуется с известными данными. В то же время мы не обнаружили значимых различий в частоте ИС у отцов и матерей пробандов.
Сегрегационный анализ проводили на основе двух способов задания дихотомического признака: фенотип А и фенотип В.
Фенотип А. Все члены родословных, имеющие сколиоз I—IV степени, считались больными. Те особи, у которых сколиоз не был выявлен при компьютерной диагностике или во время интервьюирования с родственниками, рассматривались как здоровые. Фенотип всех особей, о наличии или отсутствии сколиоза у которых не было информации, считался неизвестным.
Фенотип В. Больными считали только тех членов родословных, которые имели II—IV степень ИС. Тех, кто не имел сколиоза или имел первую степень этой патологии, считали здоровыми. При таком определении дихотомического признака часть пробандов, имеющих сколиоз I степени, оказалась отнесена к категории здоровых, и их родословные были исключены из анализа. Анализируемая выборка в этом случае была редуцирована до 66 семей, включающих 528 особей с известным фенотипом.
В табл. 4 представлены результаты сегрегационного анализа фенотипа А, полученные с помощью различных моделей. Следует отметить, что модель So, не учитывающая половые и возрастные особенности проявления болезни, описывает эмпирические данные значительно хуже, чем модель Si, учитывающая эти особенности: X2 = 29,14; р < 0,01 для Sy
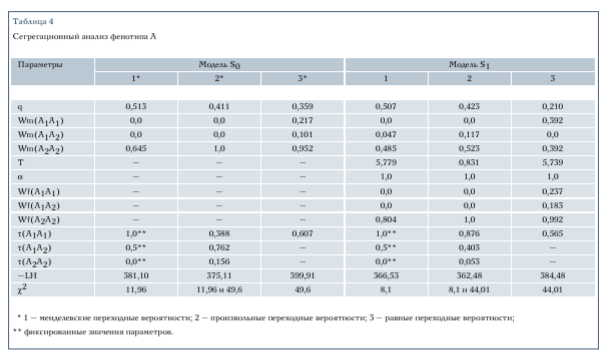
Нужно заметить, что полученные результаты не следует рассматривать как опровержение гипотезы о майор- генном контроле ИС. Существуют минимум две причины, которые могут замаскировать менделевскую сегрегацию генов и привести к ошибочному отвержению майоргенной гипотезы.
Первая из них заключается в том, что рассмотренные модели наследования признака достаточно просты. В самом деле, увеличение сложности модели привело к уменьшению разницы между гипотезами менделев- ских и произвольных переходных вероятностей: для модели S1, давшей лучшее описание эмпирических данных, критерий Эльстона - Стюарта был равен 8,1 и лишь немного превышал граничное значение х2 = 7,8; df = 3; р = 0,05. Не исключено, что дальнейшее усложнение модели может позволить продемонстрировать менделевскую сегрегацию майоргена.
К сожалению, усложнение модели всегда требует увеличения информативности эмпирических данных. Поэтому, имея конкретную выборку родословных, мы не можем тестировать на ней сколь угодно сложные модели. Вторая причина заключается в неправильном определении фенотипов у некоторых членов родословной. Было показано, что ошибочная диагностика болезни у одного члена родословной может привести к маскировке менделевской сегрегации генов [18]. Мы уже отмечали трудности, связанные с дифференциальной диагностикой ранних стадий ИС, приводящие к гипердиагностике этого заболевания. Поэтому при анализе фенотипа А в категорию больных заведомо попала часть с деформациями позвоночника, отличными от ИС. Чтобы избежать проблем с гипердиагностикой ИС, сформировали фенотип В, задав его таким образом, что все особи, имеющие слабо выраженные деформации позвоночника, были отнесены к категории здоровых.
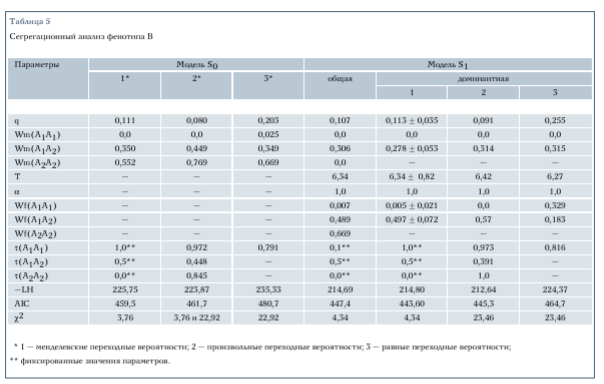
В табл. 5 приведены результаты сегрегационного анализа фенотипа В, полученные с помощью разных моделей. В этом случае даже самая простая модель S0 подтвердила менделев- скую сегрегацию генов: гипотеза с произвольными переходными вероятностями значимо не отличалась от менделевской гипотезы (х2 = 3,76). Напротив, гипотеза с произвольными переходными вероятностями была существенно лучше гипотезы с равными переходными вероятностями (X2 = 22,92; р < 0,01).
Как и в случае с фенотипом А, оказалось, что модель S0 описывает эмпирические данные значительно хуже, чем модель, учитывающая половые и возрастные особенности проявления болезни: %2 = 22,12; р < 0,01 для модели S1. Модель S1 дала примерно одинаковые оценки пенетрантностей генотипов А1А2 и А2А2. Это позволило нам предположить доминантную модель наследования фенотипа В, которая формализуется в виде W^^) = (А2А2). Как видно из табл. 3, доминантная модель значимо не отличается от более общей модели, предполагающей произвольные пенетрантности генотипов (X2 = 0,22 для S1). Согласно критерию Акайке, лучшей является доминантная модель S1, для которой МС = 443,60. Модель S1 предполагает, что возрастная зависимость проявления признака одинакова для обоих полов, а пенетрантности генотипов
различны. Для мужчин пенетрантнос- ти генотипов, содержащих мутантный аллель А2, составляют около 0,3, а для женщин - около 0,5.
Мы подтвердили менделизм для доминантной модели Sp Гипотеза с произвольными переходными вероятностями значимо не отличалась от менделевской гипотезы (X2 = 23,46; р < 0,01). Нужно отметить, что менделизм был подтвержден также при другой модели, представленной в табл. 5.
Таким образом, мы обнаружили, что наследование выраженных (II-IV степень) форм ИС можно описать в рамках доминантной майоргенной диаллельной модели с неполной пе- нетрантностью генотипов, зависящей от пола и возраста. Оценки параметров этой модели, представленные в табл. 6, позволили получить оценку распространенности выраженных форм ИС в изучаемой популяции равную 0,07-0,08. Это соответствует частоте, найденной в специальном эпидемиологическом исследовании сибирских популяций [30].
Согласно нашим данным, пенет- рантность генотипов, несущих мутантный аллель, вдвое выше у девочек, чем у мальчиков. Это хорошо соответствует известным данным о частоте ИС у детей [5]. Таким образом, полученные оценки генетических параметров хорошо согласуются с известными фактами. Это можно рассматривать как подтверждение правильности наших заключений.
Заметим, что, согласно нашей модели, ИС II-IV степени не возникает при отсутствии мутантного аллеля (пенетрантность генотипа А1А1 равна нулю). Это скорее всего указывает на генетическую гомогенность выраженных форм болезни, которая наблюдается у носителей мутантного аллеля.
Пенетрантности мутантных генотипов достаточно низки (0,3 у мальчиков и 0,5 у девочек). Однако здесь явно имеет место недооценка пенет- рантностей, так как мы полностью исключили из рассмотрения все слабо выраженные деформации позвоночника. В общем, интерпретация моделей с неполной пенетрантнос- тью генотипов достаточно сложна. Неполная пенетрантность может быть следствием модифицирующего действия других генов. Наши данные можно рассматривать лишь как указание на существование майоргена, контролирующего развитие ИС. Чтобы доказать это, надо прежде всего локализовать этот ген на одной из хромосом, то есть найти генетические маркеры, сцепленные с ним. Конечной ступенью должна быть идентификация предполагаемого гена.
В данной работе предпринималась попытка рассмотреть в качестве гена- кандидата структурный ген основного компонента внеклеточного матрикса - протеогликана. Самым крупным из протеогликанов внеклеточного матрикса является агрекан, молекула которого существует в виде агрегатов, состоящих из гиалуроновой кислоты, корового белка и присоединяющихся к нему гликозаминогликанов (хондроитинсульфатов и кератан- сульфатов). Ген AGC1, кодирующий белковый кор молекулы, локализован в 15я2б-районе. Этот ген занимает 60 тпн и состоит из 18 зкзонов, подвергающихся альтернативному сплайсингу [30].
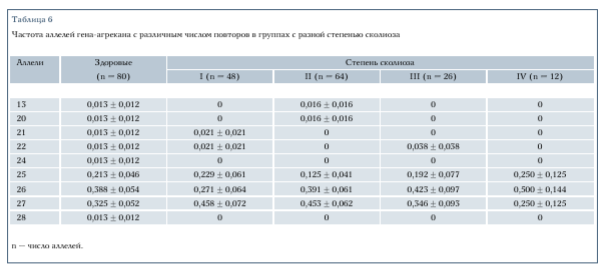
В анализируемой выборке обнаружено девять аллелей гена-агрекана, отличающихся числом тандемных повторов. Размер повторяющихся последовательностей варьировал от 775 пн для 13 повторов до 1630 пн для 28 повторов. Распределение частот аллелей в выборке среди больных и здоровых приведено в табл. 6. Здесь больными считали людей с выраженной формой ИС (II-IV степени), а все остальные были включены в группу здоровых.
Сравнение количественного состава групп, характеризуемых присутствием различных аллелей у больных и здоровых представителей родословных, показало, что эти распределения значимо не отличаются друг от друга (х2 = 5,33; df = 8; h = 0,5). Для сравнения распределений аллелей у больных и здоровых индивидов обычно используют независимо полученные случайные выборки. В нашей выборке все здоровые являются родителями детей с ИС и, следовательно, могут быть носителями аллелей, ассоциированными с болезнью, поскольку, согласно предложенной нами модели, пенетрант- ность мутантных генотипов достаточно низка. Возможно, поэтому мы не обнаружили различий в распределении аллелей среди больных и здоровых членов родословных.
Ранее было показано, что число тандемных повторов в гене-агрекана может коррелировать с тяжестью заболевания [22]. В табл. 6 показаны частоты аллелей у разных людей с разной степенью заболевания. Как видно, в исследуемой выборке не наблюдается значимых различий в распределении аллелей в различных группах (х2 =1,81; df = 8; h > 0,9). Это свидетельствует о том, что число цепей хондроитинсульфатов, присоединенных к белковому кору, непосредственно не определяет тяжесть заболевания.
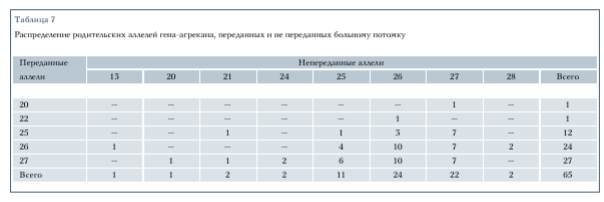
Для анализа аллельных ассоциаций с помощью TDT-теста необходимо построить распределения родительских аллелей, переданных и не переданных больному потомку. Эти распределения для анализируемой выборки представлены в табл. 7. Значение TDT-статистики, учитывающей полиаллельность изучаемого гена, посчитанное по формуле (1), равно 7,89 (df =8; h > 0,4). Как видно, оно не позволяет отвергнуть гипотезу о независимой передаче родительских аллелей больным и здоровым потомкам. Нужно заметить, что при большом числе аллелей возрастает число степеней свободы используемого критерия, что уменьшает мощность. Поэтому было решено провести ассоциацию болезни с каждым из часто встречающихся в выборке аллелей. Как видно из табл. 6, среди присутствующих в выборке аллелей наиболее часто встречаются аллели с числом повторов 25, 26, 27. Для каждого из этих аллелей была проведена неравновесность по сцеплению с помощью TDT-статистики, определенной формулой (2). Значения TDT оказались равными 0,043 (р > 0,8), 0 (p = 1) и 0,714 (р = 0,39) соответственно, что не позволяет говорить об ассоциации ИС с каким-либо из этих аллелей и подтверждает результат, полученный методом TDT.
Можно предположить несколько моделей, объясняющих участие гена- агрекана в контроле ИС. Первая из них предполагает, что причина болезни заключается в появлении аллеля гена-агрекана с аномальным числом тандемных повторов в экзоне G3. При справедливости этой модели мы должны были бы обнаружить этот мутантный аллель практически у всех больных и незначительного числа здоровых людей. Но не обнаружили заметного преобладания ни одного из аллелей в группе больных, что позволяет исключить изменение числа повторов в экзоне G3 из числа непосредственных причин развития ИС.
Вторая модель предполагает, что число цепей хондроитинсульфа- та, определенное числом тандемных повторов в экзоне G3 гена-агрекана, является не основной причиной, а одном из факторов риска развития ИС, модифицирующим действие майор- гена. В этом случае то, что мы не обнаружили ассоциации, свидетельствует о том, что эффект этого фактора риска слишком мал, чтобы быть значимым в изучаемой нами выборке.
Еще одна модель заключается в том, что мутация, вызывающая ИС, находится на участках гена-агрекана, отличных от экзона G3. В этом случае полиморфизм экзона G3 можно использовать только в качестве генетического маркера при анализе сцепления. Анализ ассоциаций аллелей эк- зона G3 и ИС мог бы дать положительный ответ только в том случае, если вызывающая болезнь мутация встречалась в составе определенного гаплотипа, объединяющего мутантный аллель и один из аллелей экзона G3. Вероятность такой ситуации в неродственных семьях, взятых из большой открытой популяции, крайне низка.
В ходе проделанной работы мы не обнаружили достоверной ассоциации ИС с полиморфизмом экзона G3, что нельзя трактовать как доказательство непричастности всего гена-агрекана к детерминации развития ИС. Чтобы проверить причастность других районов этого гена в контроле ИС, необходимо провести анализ сцепления генов. Использованный нами материал не позволяет сделать такую проверку, поскольку для анализа сцепления в отсутствии неравновесия по сцеплению необходимы родословные более сложной структуры, чем тройки, состоящие из больного потомка и его родителей [25]. В настоящее время мы собираем материал по расширенным родословным, чтобы проверить, не находится ли ген, детерминирующий ИС, вблизи экзона G3 гена-агрекана.
Выводы
ИС - генетически зависимая, контролируемая майоргеном патология, наследуемая по аутосомно-до- минантному типу с неполной пе- нетрантностью генотипов, зависящей от пола и возраста.
ИС II-IV степени не возникает при отсутствии мутантного аллеля, что свидетельствует о генетической гомогенности выраженных форм ИС, то есть дети с прогрессирующими формами ИС являются носителями мутантного гена.
Стабильные и медленно прогрессирующие формы следует отнести к функциональным деформациям позвоночника с соответствующими клиническими рекомендациями.
Исследование причастности гена- агрекана к детерминации развития ИС на малой выборке ядерных родословных не обнаружило достоверной ассоциации ИС с полиморфизмом экзона G3. В настоящее время продолжаются исследования других экзонов гена-агрека- на на расширенных родословных.
Литература
Аксенович Т.И, Гинзбург ЭХ Система для менде- левского анализа альтернативных признаков (МАН-А1) // Генетика. 1987. Т. 23. № 2. С. 268-273.
Аксенович ТИ, ЗайдманАМ, Зоркольцева И.В. идр. Новые модели наследования сложных признаков и их исследования при сегрегационном анализе идиопатического сколиоза // Генетика. 1999. Т 35. № 2. С. 255-262.
Аксенович ТИ, Семенов ИР., Гинзбург ЭХ и др. Предварительный анализ наследования сколиоза // Генетика. 1988. Т 24. № 11. С. 2056-2063. Зайдман А.М., Соловьева НА., Бородин П.М.
Клинико-генетические и морфологические исследования сколиотической болезни // Бюллетень Сибирского отделения АМН СССР. 1982. № 6. С. 82-87.
Трофимович ЕМ, Садовой М.А. Гигиеническая профилактика неинфекционных заболеваний в популяции человека // VIII Всероссийский съезд гигиенистов и санитарных врачей: Сб. науч. тр. М., 1996. Т 1. С. 186-187.
Казьмин А.И, Кон ИИ, Беленький В.Е. Сколиоз. М., 1981.
Корочкин ЛИ Взаимодействие генов в развитии. М., 1977.
Axenovich TJ, Zaidman AM, Zorkoltseva I.V, et al Segregation analysis of idiopathic scoliosis: demonstration of a major gene effect // Am. J. Med. Genet. 1996. Vol. 86. P. 389-394.
Bonaiti C., Feingold J, Briard M.L, et al. Genetics of idiopathic scoliosis // Helv. Paediatr. Acta. 1976. Vol. 31. P 229-240.
Carr AJ, Ogilvie D.J., Wordsworth B.P., et al. Segregation of structural collagen genes in adolescent idiopathic scoliosis // Clin. Orthop. 1992. N 274. P 305-310.
Chan V., Fong G.C, Luk K.D, et al. A genetic locus for adolescent idiopathic scoliosis linked to chromosome 19p13.3 // Am. J. Hum. Genet. 2002. Vol. 71. P 401-406.
Cobb J.R. Outline for the study of scoliosis. // American Academy of Orthopaedic Surgeons Instructional Course Lectures. 1948. Vol. 5. P 61-265.
Cowell HR., Hall JN, MacEwen G.D. Genetic aspects of idiopathic scoliosis. A Nicholas Andry Award essay, 1970 // Clin. Orthop. 1972. N 86. P. 121-131.
Elston R.C. Segregation analysis // In: Harris H., Hirschhorn K., eds. Advances in Human Genetics. N.Y., 1981. Vol. 11. P. 63-120.
Elston R.C., Stewart JA General model for the genetic analysis of pedigree data // Hum. Hered. 1971. Vol.21. P. 523-542.
Giampietro P.F., Raggio C.L., Blank R.D. Synteny- defined candidate genes for congenital and idiopathic scoliosis // Am. J. Med. Genet. 1999. Vol. 83. P. 164-177.
Goldberg C.J., Dowling F.E., Fogarty E.E., et al. Adolescent idiopathic scoliosis and cerebral asymmetry. An examination of nonspinal perceptual system // Spine. 1995. Vol. 20. Р. 1685-1691.
Hodge S.E., Greenberg DA Sensitivity of lod scores to changes in diagnostic status // Am. J. Hum. Genet. 1992. Vol. 50. P. 1053-1066.
Horton WE., Lethbridge-Cejku M., Hochberg M.C, et al.
An association between an aggrecan polymorphic allele and bilateral hand osteoarthritis in elderly white men: data from the Baltimore Longitudinal Study of Aging (BLSA) // Osteoarthritis Cartilage. 1998. Vol. 6. P. 245-251.
Inoue M., Minami S., Nakata Y., et al. Prediction of curve progression in idiopathic scoliosis from gene polymorphic analysis // Res. into Spin. Deform. 2002.Vol. 4. P. 90-96.
Justice CM, Miller NH, Marosy B., et al. Familial idiopathic scoliosis: evidence of an X-linked susceptibility locus // Spine. 2003. Vol. 28. P. 589-594.
Kawaguchi Y., Osada R., Kanamori M., et al. Association between an aggrecan gene polymorphism and lumbar disc degeneration // Spine. 1999. Vol. 24. P. 2456-2460.
Miller NH, Mims B., Child A., et al. Genetics analysis of structural elastic fiber and collagen genes in familial adolescent idiopathic scoliosis // J. Orthop. Res. 1996. Vol. 14. P. 994-999.
Neyman J., Pearson E.S. On the use and interpretation of certain test criteria for the purposes of statistical inference // Biometrica. 1928. Vol. 20A. P. 175-263.
Ott J. Analysis of Human Genetic Linkage. 3rd edition. Baltimore; L., 1999.
Salehi L.B., Mangino M., De Serio S., et al. Assignment of a locus for autosomal dominant idiopathic scoliosis (IS) to human chromosome 17p11 // Hum. Genet. 2002. Vol. 111. P 401-404.
Sham P.C. Statistics in Human Genetics. N. Y.; Toronto, 1998.
Spielman R.S, Ewens WJ. The TDT and other family- based tests for linkage disequilibrium and association // Am. J. Hum. Genet. 1996. Vol. 59. P 983-988.
Stokes IA, Bigalow L.C. Moreland M.S. Measurement of axial rotation of vertebrae in scoliosis // Spine. 1986. Vol. 11. P 213-218.
Watanabe H, Yamada Y.,Kimata K.Roles of aggrecan, a large chondroitin sulfate proteoglycan, in cartilage structure and function // J. Biochem. 1998. Vol. 124. P. 687-693.
Wise CA, Barnes R, Gillum J, et al. Localization of susceptibility to familial idiopathic scoliosis // Spine. 2002. Vol. 25. P 2372-2380.
Wynne-Davies R. Familial idiopathic scoliosis. A familial survey // J. Bone Joint Surg. Br. 1968. Vol. 50. P. 24-30.
Wynne-Davies R. Genetic aspects of idiopathic scoliosis. // Dev. Med. Child. Neurol. 1973. Vol. 15. P809-811.
.М. Зайдман - Новосибирский НИИ травматологии и ортопедии
Т.И. Аксенович - Институт цитологии и генетики СО РАН, Новосибирск
М.А. Садовой - Новосибирский НИИ травматологии и ортопедии
И.Л. Трегубова - Новосибирский НИИ травматологии и ортопедии
Р.Н. Шарипов - Институт цитологии и генетики СО РАН, Новосибирск
Теги: идиопатический сколиоз
234567 Начало активности (дата): 11.08.2024 21:17:00
234567 Кем создан (ID): 989
234567 Ключевые слова: идиопатический сколиоз, наследственность, генотип, фенотип, майорген, экзон, агрекан
12354567899
Похожие статьи
Результат использования корсета 3D немецкой школы в лечении пациентки с ювенильным идиопатическим сколиозомРентген на дому 8 495 22 555 6 8
МСКТ-семиотика позвонков у больных со стенозом шейного отдела позвоночника
Анализ заболеваемости костно-мышечной системы у детей и организация специализированной помощи в Санкт-Петербурге
Шейный отдел позвоночника при болезни Шойермана: обзор литературы


